1
GIỚI THIỆU LUẬN ÁN
1. Tính cấp thiết của đề tài
Bệnh thalassemia thuộc nhóm bệnh rối loạn tổng hợp huyết sắc tố, là bệnh di
truyền phổ biến nhất trên thế giới với ước tính khoảng 7% dân số mang gen bệnh.
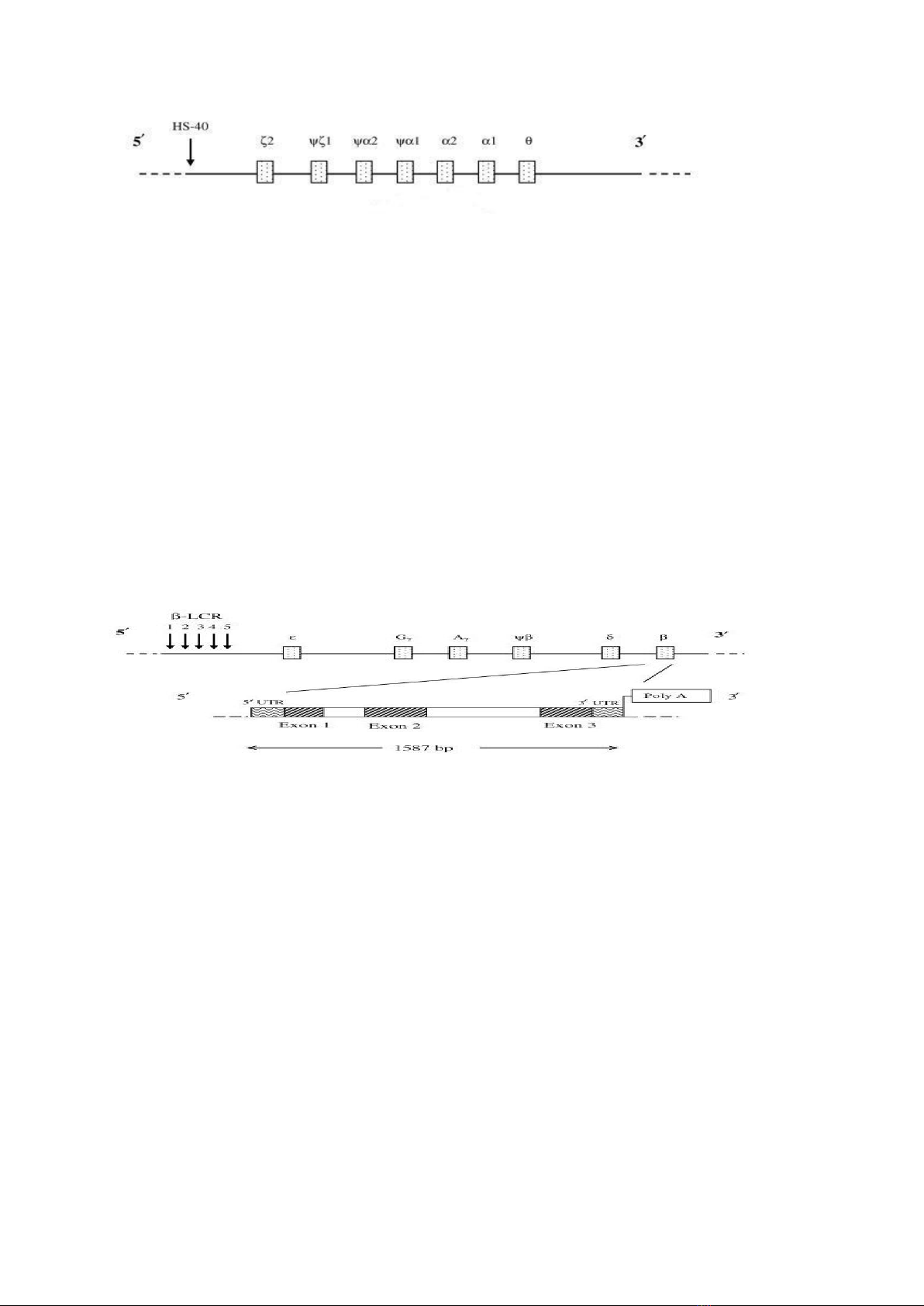
Nguyên nhân gây bệnh là do đột biến gen quy định tổng hợp chuỗi globin dẫn đến
mất cân bằng các loại chuỗi globin, tạo nên bất thường về huyết sắc tố, làm giảm
chất lượng hồng cầu, gây ra tình trạng thiếu máu ở bệnh nhân. Vì thiếu máu, cơ thể
tăng hấp thu sắt và tăng tích lũy tại tổ chức trong cơ thể do phải truyền máu, từ đó
gây ra rất nhiều biến chứng tại gan, tim, tuyến nội tiết làm giảm chất lượng cuộc
sống và tuổi thọ của người bệnh.
Thalassemia là bệnh phòng được và chữa được. Phòng bệnh thalassemia là
phòng không ra những trẻ bị bệnh. Để phòng bệnh, nhất thiết phải biết chính xác đột
biến và đặc điểm lâm sàng của đột biến gây bệnh thalassemia. Những người bệnh
thalassemia hoàn toàn có thể có được cuộc sống gần như bình thường nếu được
truyền máu và không bị quá tải sắt tại các tổ chức.
Ở Việt Nam, mặc dù chưa có chương trình phòng bệnh thalassemia quốc gia.
Năm 2016, bộ Y tế đã ban hành văn bản hướng dẫn chẩn đoán điều trị bệnh
thalassemia, trong đó đã qui định việc chẩn đoán xác định, theo dõi điều trị quá tải
sắt ở bệnh nhân thalassemia và thực hiện phòng bệnh thalassemia.
2. Mục tiêu của đề tài
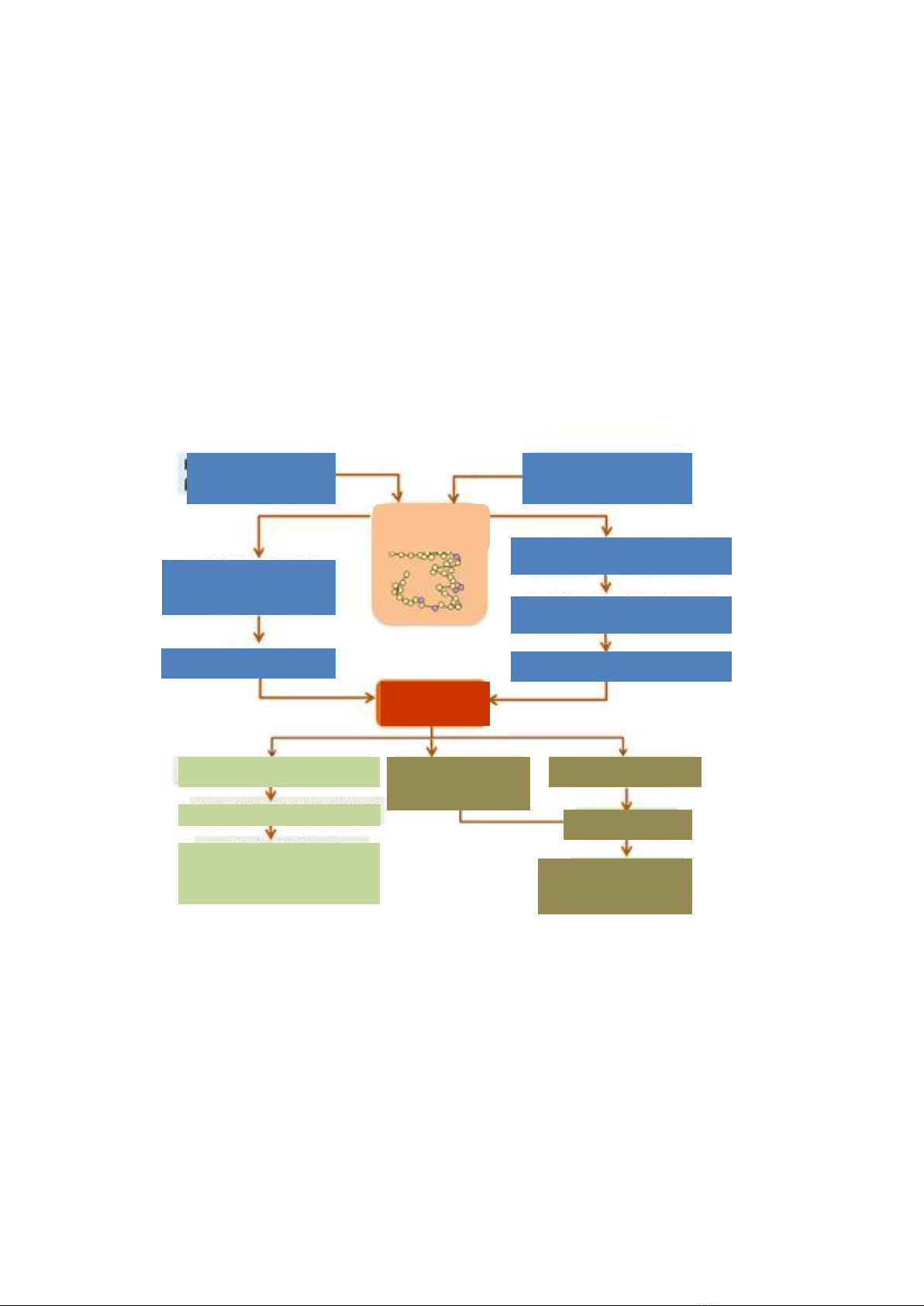
1. Nghiên cứu đặc điểm đột biến gen globin bằng kỹ thuật Strip Assay tại Viện
Huyết học - Truyền máu Trung ương giai đoạn 2013 - 2016;
2. Đánh giá sự thay đổi một số chỉ số quá tải sắt bằng MRI ở bệnh nhân
thalassemia được điều trị thải sắt.
3. Ý nghĩa thực tiễn và đóng góp mới của đề tài
Đóng góp mới về khoa học:
- Triển khai, ứng dụng thành công kỹ thuật Globin Strip Assay trong chẩn đoán
đột biến gen globin cho các đối tượng có mang gen bệnh thalassemia;
- Ứng dụng thành công MRI để chẩn đoán chính xác tình trạng quá tải sắt (QTS)
tại gan, tim để từ đó tiên lượng biến chứng tại các tổ chức gan, tim và nội tiết ở BN
thalassemia.
Giá trị thực tiễn của đề tài:
- Việc xác định được đột biến gen globin và đặc điểm lâm sàng của các đột biến
rất có ý nghĩa trong chẩn đoán và đặc biệt là phòng bệnh thalassemia;
- Chẩn đoán mức độ quá tải sắt tại gan, tim ở BN thalassemia từ đó đưa ra phác
đồ điều trị phù hợp và tư vấn cho BN kịp thời để ngăn chặn biến chứng nặng nề, góp
phần nâng cao chất lượng cuộc sống cho người bệnh.
4. Cấu trúc luận án