
Determination of the consensus binding sequence for the purified
embryonic heat shock factor 2
Martine Manuel
1,
*
,
†, Murielle Rallu
1,
*
,
‡, Marie-The
´re
`se Loones
1
, Vincenzo Zimarino
2
, Vale
´rie Mezger
1
and Michel Morange
1
Laboratoire de Biologie Mole
´culaire du Stress, Unite
´de Ge
´ne
´tique Mole
´culaire UMR8541, Ecole Normale Supe
´rieure, Paris, France;
2
DIBIT, San Raffaele Scientific Institute, Milan, Italy
Heat shock transcription factors (HSFs) are characterized
by their ability, upon activation, to bind to heat shock
response elements (HSE) present in the promoter of their
target genes. HSE are composed of inverted repeats of the
pentamer nGAAm. In this study, we compare the
embryonic HSF2 protein, purified from F9 embryonal
carcinoma cells tumor, and the in vitro synthesized HSF2.
We show that the context of HSF2 synthesis influences its
thermosensitivity and DNA-binding properties. Therefore,
we determined the consensus binding sequence for the
purified embryonic HSF2 by the technique of systematic
evolution of ligands by exponential enrichment (SELEX).
We show that embryonic HSF2 prefers sites containing
three or four nGAAm inverted pentamers and that its
optimal binding sequence contains the 8-mer palindromic
core 5¢-TTCTAGAA-3¢. The consensus binding sequence
for the embryonic HSF2 will be very helpful to identify
new targets for this factor, during developmental and
differentiation processes.
Keywords: heat shock transcription factor-2; protein purifi-
cation; cooperativity; SELEX; consensus binding sequence.
Heat shock factor 2 (HSF2) belongs to the vertebrate heat
shock factor family that also includes HSF1, HSF3 and
HSF4 [1–5]. The members of the HSF family are defined by
their ability to specifically bind the regulatory sequence heat
shock element (HSE) [6]. Located in the regulatory regions
of heat shock genes, HSE consists of the inverted repeat of a
basal element nGAAm [7]. Two inverted repeats are
sufficient for Drosophila HSF binding, but optimal binding
is obtained with three repeats [8]. In agreement with this
observation, the activated form of HSFs has been demon-
strated to be a trimer in yeast [9], in Drosophila [10], in
human [11,12] or in mouse [13]. The HSE-binding activity of
heat shock factors is not constitutive, but induced by
various stresses, by differentiation or developmental pro-
cesses. HSF1 and HSF3 are activated by stresses that elicit
the so-called heat shock responseand induce the tran-
scription of heat shock genes. HSF1 corresponds to the
paradigm member of the family and is the functional
homolog, for its function in the heat shock response, of the
unique HSF found in yeast and Drosophila. Avian HSF3 is
activated by more severe stresses than HSF1, but is also
required for an optimal response to stress [14,15]. Indeed,
avian cells expressing HSF1, but in which the HSF3 gene
has been disrupted, exhibit a diminished response to stress,
even at mild heat shock temperatures [14]. Athough
heterotrimers were never detected, HSFs may interact with
each other in a more complex way.
HSF4 is an exception and constitutively binds DNA as a
trimer in the absence of stress. Its expression is regulated in a
tissue-specific manner [5,16]. The Hsf4 gene generates both
an activator or a repressor of heat shock genes by alternative
splicing; the tissue-specificity of the two forms may create a
modulation of expression of hsps in the different tissues.
In contrast to HSF1 and HSF3, HSF2 is not activated in
response to heat shock or other cellular stresses. It is found
in a trimeric DNA-binding form during hemin-induced
differentiation of the human erythroleukemia cells K562, in
mouse embryonal carcinoma (EC) cells, and during mouse
embryogenesis and spermatogenesis. During the differenti-
ation of K562 cells, HSF2 is converted from an inert dimeric
form to a DNA-binding trimer that is able to induce the
transcription of Hsp70 gene [17–19]. In this system, it seems
that although HSF1 and HSF2 are activated by distinct
signals, they also induce a similar profile of heat shock gene
transcription [17,18]. It was therefore suggested that in
mammalian cells, HSF1 was responsible for heat shock gene
induction upon stress, while HSF2 was responsible for the
high spontaneous expression of heat shock genes, which is
observed in the absence of stress in EC cells, and during
mouse embryogenesis and spermatogenesis.
However, an accumulation of data shows that the
contribution of HSF2 to the transcriptional regulation of
heat shock genes remains unclear. Indeed, athough HSF2
Correspondence to M. Morange, Laboratoire de Biologie Mole
´culaire
du Stress, Unite
´de Ge
´ne
´tique Mole
´culaire UMR8541, Ecole Normale
Supe
´rieure, 46 rue d’Ulm, 75230 Paris cedex 05, France.
Fax: + 33 1 44 32 39 41, Tel.: + 33 1 44 32 39 46,
E-mail: morange@wotan.ens.fr
Abbreviations: HSF, heat shock transcription factor; HSE, heat shock
response elements; SELEX, systematic evolution of ligands by
exponential enrichment; EC, embryonal carcinoma; in vitro
synthesized, i.v.s.
*Note: these authors contributed equally to this work.
Present address: Department of Biomedical Sciences,
University of Edinburgh, UK.
Present address: Developmental Genetics Program, Skirball Institute
for Biomolecular Medicine, NYU Medical Center, New York, USA.
(Received 5 December 2001, revised 28 February 2002,
accepted 5 April 2002)
Eur. J. Biochem. 269, 2527–2537 (2002) FEBS 2002 doi:10.1046/j.1432-1033.2002.02917.x

displays a strong DNA-binding activity in EC cells [20,21],
the HSE region of Hsp70 promoter was found unoccupied
by HSF2 [21]. Controversial data suggest that, in contrast to
what was observed in mouse, HSF2 does not display any
DNA-binding activity at any stage of the rat seminiferous
epithelial cycle and that HSF2 expression does not correlate
with any HSP expression pattern [22]. No correlation is
found during pre- or post-implantation embryogenesis
between the expression patterns of major HSPs and HSF2
profiles [23]. Even in the case of the K562 cell system, where
the HSE sites were found occupied in vivo by HSF2 during
hemin-induced differentiation [18], other data suggest a role
of HSF1 and not HSF2 in the hemin-induced transcription
of Hsp70 gene [24], re-addressing the respective role of the
two factors in Hsp70 expression during differentiation.
Therefore, the role of HSF2 during differentiation and
development is likely distinct from a simple inducer of heat
shock genes in nonstress conditions, in differentiation or
developmental situations. Its role is still not unravelled and
its targets as a transcription factor unknown.
Studies performed on recombinant HSF1 and HSF2,
produced in E. coli, using random oligonucleotide selection
have shown that they display slightly distinct preferences,
although both factors bind to the 5¢-nGAAm-3¢basal motif
[25]. Recombinant HSF2, in contrast to HSF1, does not
bind to HSE in a cooperative manner. We purified HSF2
from F9 mouse embryonal carcinoma tumors and analyzed
its DNA-binding properties at various temperatures in
comparison with in vitro synthesized (i.v.s.) HSF2 protein,
produced in reticulocyte lysates. This study demonstrates
that the DNA-binding properties of the purified HSF2 are
different from those of the i.v.s. HSF2. This suggests that
HSF2 function is highly sensitive to the environment in
which it is synthesized. We therefore decided to determine
the consensus binding sequence for the purified embryonic
factor, by a SELEX assay using a semirandom oligonucle-
otides library. We found that the embryonic factor requires
at least three 5¢-nGAAm-3¢motifs and that its optimal
binding sequence contains a palindromic 8-mer core
5¢-TTCTAGAA-3¢. This result is in contrast to what was
found for the recombinant HSF2.
MATERIALS AND METHODS
Oligonucleotides
The oligonucleotides used in this study are shown in Table 1.
Embryonal carcinoma (EC) cell culture, acquisition
of tumors in 129 mice and purification of HSF factors
F9 EC cells were grown and extracts were prepared as
previously described [20]. F9 tumor cells were obtained by
subcutaneous injection of 2 ·10
6
F9 cells in 5-week-old
syngenic mice (strain 129). Tumors were allowed to grow for
about 2 weeks. After cervical dislocation, the tumors were
rapidly dissected and immediately frozen in dry ice until use
for extraction. Appropriate measures were taken to minim-
ize animals pain or discomfort, in accordance with the
European Communities Council Directive of 24 November
1986 (86/609/EEC).
For HSF2 protein purification, 26 g of crude material
(18 tumors) were extracted with 300 mL of extraction
buffer (10 m
M
Hepes pH 7.9, 0.4
M
NaCl, 0.1
M
EGTA,
0.5 m
M
dithiothreitol, 5% glycerol, 0.5 m
M
phenyl-
methanesulfonyl fluoride supplemented with 1 lgÆmL
)1
pepstatin and 1 lgÆmL
)1
aprotinin). Whole-cell extracts
were clarified by centrifugation for 30 min at 100 000 g
and the supernatants were stored at )80 C. The final
protein concentration of the extracts averaged
5.8 mgÆmL
)1
. The complete purification of HSF2 protein
was performed by adaptation of a three-step protocol
previously described by Wu et al.[26].
(a) Whole-cell F9 tumor extracts were applied on an
heparine-sepharose column (CL-6B, Pharmacia), washed
with 300 mL of equilibration buffer (0.15
M
NaCl, 20 m
M
Hepes pH 7.9, 0.1 m
M
EGTA, 10% glycerol, 0.5 m
M
PMSF, 1 lgÆmL
)1
pepstatin and 1 lgÆmL
)1
aprotinin).
Bound proteins were eluted with a linear salt gradient
from 0.15 to 1.5
M
NaCl. Fractions were analyzed by
electromobility shift assay (EMSA) and those containing
an HSE-binding activity (0.2 to 0.6
M
NaCl) were pooled.
The yield and purification factors were calculated for this
column and were found to be equal to 87% and 5.6,
respectively.
(b) A DNA-affinity resin was prepared by coupling
HSE sequences to a CNBr-activated sepharose (CL-4B;
Pharmacia Biotech), according to Kanodaga and Tjian
[27]. The synthetic HSE oligonucleotide CTAGAAGCTT,
similar to that of Sorger & Pelham [28], was annealed with
itself in order to form double-stranded molecules with
protruding ends, which were subsequently ligated. This
resulted in the formation of polymers of about
100–200 bp, as estimated by agarose gels, that were linked
to the resin. Fractions containing the HSE-binding activity
were pooled and adjusted, by dilution, to 0.35
M
NaCl,
26 m
M
Hepes pH 7.6, 20% glycerol, 0.3 m
M
dithiothrei-
tol, 0.5 m
M
phenylmethanesulfonyl fluoride. The diluted
fractions were incubated overnight at 4 C, under gentle
agitation, in the presence of resin, protease inhibitors
(1 lgÆmL
)1
pepstatin and aprotinin) as well as
1.5 lgÆmL
)1
poly(dI-dC).poly(dI-dC) to avoid nonspecific
interactions. After extensive washing with 0.2
M
NaCl
equilibration buffer (26 m
M
Hepes pH 7.6, 20% glycerol,
0.1% NP40, 0.1 m
M
EGTA, 0.3 m
M
dithiothreitol,
Table 1. Oligonucleotide sequences used in this study.
HSE2 5¢-TCGACAGATCTCCTAGAACGTTCTAGA
AGCTTCGAGAGGATTC-3¢
2U519m 5¢-CAGAATCTTCTCGATAGTTAGG-3¢
SHVAL 5¢-CTAGAACGTTCTAGAAGCTTCGAGA-3¢
SHVAL-SPZ 5¢-CTAGAACGTTCTAGAGAGTTTCCAG-3¢
NOG 5¢-CTAGAACGTTCTAGGGGGGGGGG-3¢
NOA 5¢-CTAGAACGTTCTAAAAAAAAAAA-3¢
MTH 5¢-CTAGAACGTTCTAAAAATTTCCAG-3¢
MCL 5¢-CTAGAACGTTCTAAAAAATTTCCAG-3¢
SHC 5¢-CTAGAACGTTCTAGAGAGAGAGAGA-3¢
JUL 5¢-CTAGAACGTTCTAGAACGTTCTCA-3¢
Deg-sb 5¢-CACGTGCGCTGGTACN
3
GAANNTTC
N
14
GGCTATCGACTGGCG-3¢
CL39 5¢-ATGGAACATTCTAGAACCTTCTCTT-3¢
CL83 5¢-AGAGAACATTCTAGAACATGGGTAC-3¢
83woTA 5¢-AGAGAACATTCACGAACATGGGTAC-3¢
39woGAA 5¢-ATGCACCATTCTAGAACCTTCTCTT-3¢
83woGAA 5¢-AGACACCATTCTAGAACATGGGTAC-3¢
2528 M. Manuel et al. (Eur. J. Biochem. 269)FEBS 2002

0.5 m
M
phenylmethanesulfonyl fluoride, 1 lgÆmL
)1
pepst-
atin and aprotinin), proteins specifically bound to the resin
were eluted by steps of increasing NaCl concentration
(0.3 to 2
M
). Fractions were analysed by EMSA and those
containing HSE-binding activity (0.6 to 1.6
M
NaCl) were
pooled, concentrated 20-fold and analyzed by SDS/
PAGE. Silver staining of the gel revealed the presence
of several bands.
(c) Therefore, active fractions were subjected to a second
DNA-affinity chromatography. Fractions from the first
affinity chromatography were brought a second time to
0.35
M
NaCl, incubated with the HSE-affinity resin and
eluted exactly as before. Elution of HSE-binding activity
occured between 0.6 and 1.7
M
NaCl. The active fractions
were pooled and used for gel-shift assay.
During the two successive steps of DNA-affinity chro-
matography, fractions were collected in silanized tubes to
prevent sticking on plastic walls. Total yields in HSE-
binding activity and protein amount were estimated and
allowed to calculate a total purification factor equal to 3000.
SDS/PAGE and Western-blot analysis
Fractions containing HSF2 protein were pooled and loaded
on G25 sephadex (NAP columns; Pharmacia Biotech) in
order to discard most of the salts. Eluted material from the
Sephadex columns was then lyophilized and resuspended in
water so that the final volume was 100-fold less than at the
beginning. Three quarters of this concentrated material was
loaded on a 10% polyacrylamide gel and revealed by silver
staining in parallel with known concentrations of BSA to
estimate the amounts of purified protein. The last quarter
was used for Western blotting after transfer to a nitrocel-
lulose filter. HSF2 polyclonal antibodies were used at
1 : 2500 dilution as previously described [23]. Detection was
performed using the ECL peroxydase detection system
(Amersham).
Electromobility shift assays (EMSA)
Binding reactions were performed as described previously
[20]. Both strands of the DNA template were
32
P end-
labeled using T4 polynucleotide kinase and [c-
32
P]ATP.
Fourteen microliters of extracts containing 10–20 lgof
proteins from crude extracts, 0.7 ng of pure HSF2 protein
or 3 lLofin vitro translated proteins were mixed with 9 lL
of binding solution [0.2 ng of
32
P-labeled double-stranded
DNA template, 4 lg of double stranded polydI-dC, 9% (w/
v) Ficoll, 44 m
M
Hepes pH 7.6, 2.2 m
M
MgCl
2
and 88 m
M
KCl]. In competition experiments, 20 ng of unlabeled
double stranded DNA template were added to the binding
solution. The reaction mixtures were loaded on a 4%
acrylamide gel (acrylamide/bisacrylamide, 29 : 1, w/w) in
0.25 ·Tris/borate/EDTA buffer.
Analysis of HSF2 thermosensitivity properties
HSF2 factors (i.v.s. or embryonic) were incubated at a
moderate (37 C) or high temperature (44 or 45 C) and
samples were taken at increasing periods of time, brought to
room temperature and subjected to the binding reaction in
presence of the oligonucleotide HSE2. Samples were then
immediately loaded on the migrating gel. Quantification of
the signal in the specific retarded complexes was performed
using a Bas1000 Imager (Fuji) after 1 h exposure. Arbitrary
values measured at distinct incubation times were standard-
ized to the initial value.
Multiple probes band shift assay
Synthetic oligonucleotides, containing an increasing num-
ber of the conserved 5 bp units nGAAm (organized in
contiguous arrays where each unit is inverted relative to
the immediately flanking one) and their complementary
strands were obtained from Genset (Paris, France). The
same oligonucleotides as those described by Xiao et al.
[29] were used, where n and m are A and T, respectively,
for GAA and TTC. Flanking sequences, added to this
core region in order to limit self-annealing, were identical
to those present at the ends of the oligonucleotide used
for affinity chromatography.According to the number of
repeats, oligonucleotides were named Rep2, Rep3, Rep4,
Rep5 and Rep6.
Binding reactions were performed as described above,
except that the binding solution contained a total amount
0.2 ng of
32
P end-labeled double-stranded oligonucleotides
corresponding to a mixture of Rep2, Rep3, Rep4, Rep5
and Rep6 at the same concentration. Protein extracts and
range of protein amounts used to perform these experi-
ments were as follows: 0.7 ng of HSF2 protein purified to
homogeneity (supplemented with 200 lgofBSA),and
3lL of recombinant HSF2 protein expressed in reticulo-
cyte lysates. Binding reactions were performed at room
temperature during increasing periods of time ranging
from 0.5 min to 3 h and were followed by pore exclusion
limit electrophoresis. Samples were loaded on a 3–10%
gradient acrylamide gel (acrylamide/bisacrylamide, 29 : 1,
w/w) and migration was performed for 6 h at 350 V in
0.25 ·Tris/borate/EDTA buffer, until the complexes
reached a position in the gel preventing their migration.
The position of specific complexes was detected by direct
autoradiography.
Bands containing the specific complexes as well as free
DNA were cut out of the gel and oligonucleotides present in
these gel slices were eluted overnight in distilled water at
37 C. Samples were extracted once in phenol-chloroform
and once in chloroform, then concentrated in speed-vacuum
apparatus. They were then directly resuspended in the
sequencing loading buffer and analyzed on a denaturing
10% polyacrylamide gel in 1x TBE. The relative amounts of
the different oligonucleotides contained in each band were
quantified as previously described.
SELEX assay
The SELEX procedure was performed according to a
strategy described previously [30].
Preparation of a random sequence library
The 55-mer oligonucleotides Deg-sb (5¢-CACGTGCGC
TGGTACN
3
GAAN
2
TTCN
14
GGCTATCGACTGGCG-
3¢), containing two inverted trimers GAA and 19 random
nucleotides, and two PCR primers: P1, corresponding to the
first (top strand) 15 bases, and P2, complementary to the
last (bottom strand) 15 bases, were manufactured by
FEBS 2002 Determining the optimal binding sequence for HSF2 (Eur. J. Biochem. 269) 2529

Eurobio (Les Ulis, France). A random sequence library,
Sel0, was generated by a primer extension reaction carried
out with Deg-sb as template and the (bottom) primer P2.
800 pmol of Deg-sb, annealed to a mix of 1600 pmol of cold
P2 and 80 pmol of radiolabeled P2, were extended with
100 U of Klenow fragment in a 200-lL Klenow reaction
mixture. The extended products were purified on a 12%
acrylamide gel.
Selection and amplification of sequences
that bind HSF2
Sel0 (450 ng in 90 lL of binding solution) was mixed with
30 mL of pooled elution fractions of purified embryonic
HSF2 (7 ng) and 1 mg of BSA. The reaction mixture
was incubated 15 min at room temperature and loaded on
a 4% acrylamide gel. After migration, the wet gel was
wrapped with Saran and exposed to X-ray film. The gel
region harboring HSF2-Sel0 complexes was localized by
comparison with the electrophoretic mobility of the
HSF2-radiolabeled Shvalspz complex that was loaded
on the adjacent control lane. An appropriate gel slice was
excised (large enough to take into account the smeary
binding pattern displayed by the purified HSF2 protein)
and soaked overnight at 37 C in elution buffer (0.3
M
NaCl, 1 m
M
EDTA, 0.1% SDS). The eluted DNA was
purified on a Sephadex G-25 column (NAP-25 column,
Pharmacia Biotech) and concentrated to a volume of
50 lL in water. A 5-lL sample was added to a PCR
mixture together with 75 pmol of primer P1, 75 pmol of
primer P2 and 2.5 U of Tfl DNA polymerase (Promega)
in a final volume of 100 lL containing 20 m
M
Tris/acetate
(pH 9), 10 m
M
ammonium sulfate, 75 m
M
potassium
acetate, 0.05% Tween 20, 1.25 m
M
MgSO
4
,and75l
M
of each dNTP. Eight such reaction mixtures were set up.
The samples were heated for 1 min at 94 C (hot start).
For each 35 cycles of PCR, samples were denatured at
94 C for 30 s, annealed at 46 C for 30 s, and extended
at 75 C for 15 s. All eight reaction mixtures were pooled
and the DNA was purified on a 12% acrylamide gel.
About 50–100 ng of DNA was used for the next cycle of
the SELEX procedure.
Cloning of the products of selection
DNA amplified from the last cycle of selection was rendered
blunt-ended using T4 DNA polymerase and inserted at the
EcoRV site of pBluescript (pKS+, Stratagene).
Sequencing of the products of selection
After each round of selection, the amplified selected
DNA was sequenced as follows using the T7-sequencing
kit from Pharmacia Biotech with the following modifi-
cations to take into account the short size of the
sequences. P2 (10 ng) was end-labeled with [c-
32
P]ATP
and annealed to 10 ng of selected DNA in a 14-lL
volume containing 0.15
M
Tris/HCl (pH 7.6), 15 m
M
MgCl
2
and 23 m
M
dithiothreitol. The mix was boiled
for 5 min and left on ice for 10 min 4 U of T7 DNA
polymerase in 2 lL of dilution buffer [20 m
M
Tris/HCl
(pH 7.5), 5 m
M
dithiothreitol, 100 lgÆmL
)1
BSA and 5%
glycerol], 4 lLof33m
M
NaCl and 1 lLof100m
M
MnCl
2
, 150 m
M
sodium isocitrate were added to the
annealing mix on ice. 4.5 lL of this mixture were added to
2.5 lL of each of the four ddNTP Mix-Short solutions
[840 l
M
each dN
1
TP, dN
2
TP, dN
3
TP; 93.5 l
M
dN
4
TP;
14 l
M
ddN
4
TP; 40 m
M
Tris/HCl (pH 7.6) and 50 m
M
NaCl]. The reaction mix was incubated at 37 Cfor
20 min The sequences were analysed on a 10% denaturing
acrylamide gel.
Individual Sel6 clones were sequenced using the T7
sequencing kit from Pharmacia Biotech and the T7 primer
according to the manufacturer’s instructions.
RESULTS
Purification of mHSF2 protein from EC cells
Sufficient starting amounts for the purification of HSF2
protein were obtained from tumors of F9 embryonal
carcinoma cells. These tumors were produced by injection
of F9 cells, in which HSF2 is highly expressed, under the
skin of syngenic mice. We verified that extracts produced
from tumor cells displayed an HSE-binding activity similar
to that of extracts from in vitro cultivated F9 cells (data not
shown),showing that mouse or tissue manipulations did not
uncover any stress-inducible activity (due to HSF1 protein).
The complete procedure for HSF2 purification combined
heparin and DNA affinity chromatographies [26]. HSF2
protein elution profile was monitored by the presence of an
HSE-binding activity in gel-shift assay (at room tempera-
ture). The first step of this purification procedure (i.e. the
heparine–sepharose chromatography) led to the separation
of HSF2 protein from 80% of the proteins present in crude
extracts. The following steps consisted of two HSE-affinity
chromatographies (see Materials and methods). After the
first one, HSF2 protein was separated from most of the
remaining proteins but a few of them were still co-eluted
with it. Therefore, HSF2-containing fractions were reloaded
on the same column in order to obtain a pure protein.
Analysis on SDS/PAGE after silver-staining showed one
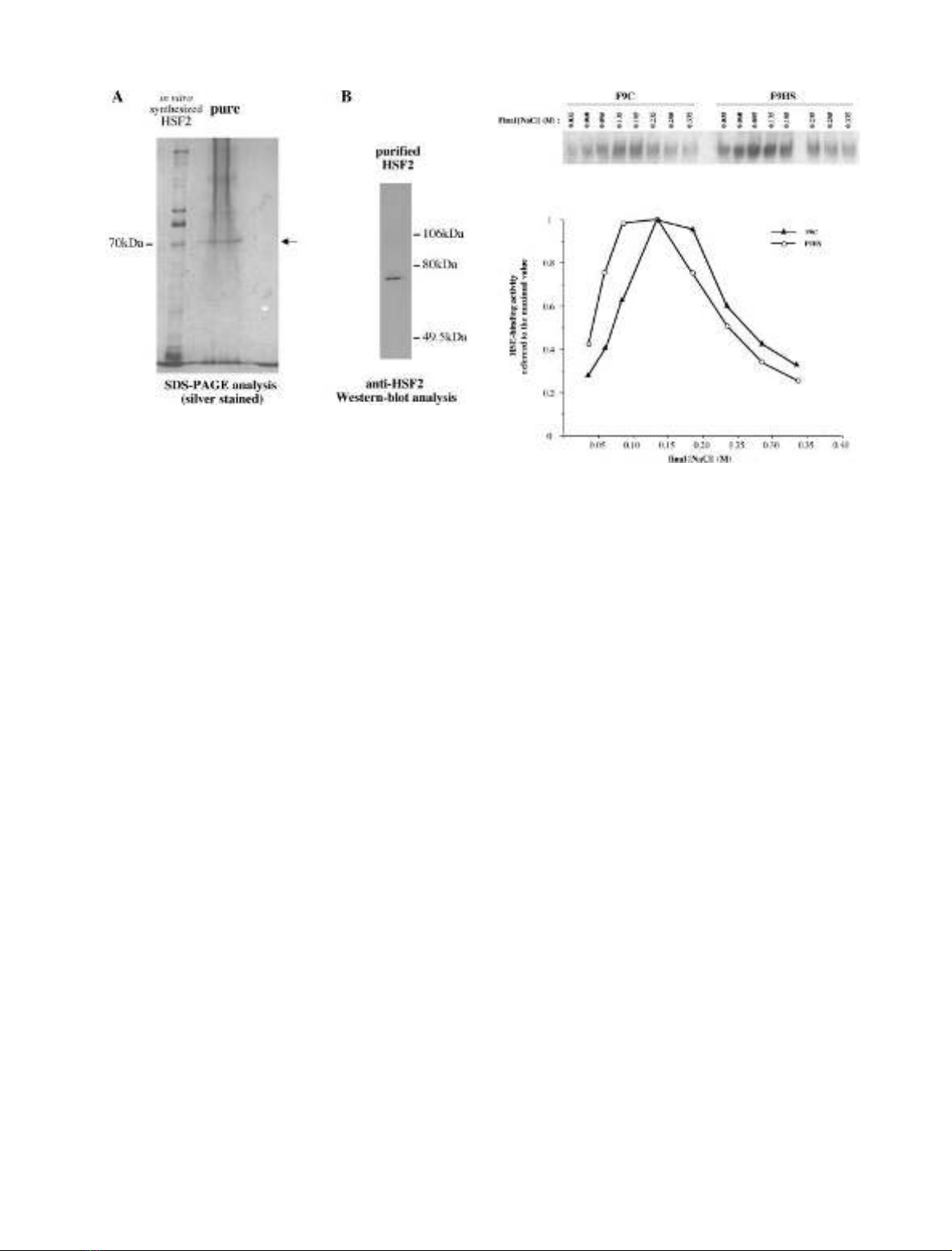
unique band of 70 kDa (Fig. 1A). This band was
recognized by HSF2 antibodies (Fig. 1B) and comigrated
with one protein product present in reticulocyte lysates
expressing HSF2 protein. Thus, it appeared that HSF2
protein from F9 embryonic cells was purified to near
homogeneity. The purification factor was estimated to be
equal to 3000.
The pure protein was stable at )70 C and could sustain
more than two cycles of freeze-thawing. However, gel shift
assays with pure protein gave poor reproducible results, and
we considered that, at these low protein concentrations, the
rare molecules of HSF2 protein might stick on the tube
walls, even when silanized. Therefore, we added 200 lgof
BSA to each point of binding reaction and got a reprodu-
cible stabilization of the purified HSF2 protein. We called
HSF2 purified from F9 tumor cells embryonic HSF2.
Conditions of binding and elution of HSF2 protein, in the
affinity column, gave several informative results about its
properties. Indeed, whereas binding conditions of HSF2
protein to the heparine–sepharose resin were similar to that
of Drosophila HSF, conditions used for the HSE-DNA
affinity chromatography were quite different. HSF2-con-
taining fractions required a longer incubation time with the
resin in order to bring the reaction to completion and the
2530 M. Manuel et al. (Eur. J. Biochem. 269)FEBS 2002
ionic strength had to be increased to 0.35
M
NaCl (in
comparison to 0.25
M
NaCl for Drosophila HSF). In fact,
we showed that optimal binding to HSE sequences occurred
at slightly higher NaCl concentrations for HSF2 protein
(present in extracts from F9 control cells) than for HSF1
(present in extracts from F9 heat-shocked cells), the
Drosophila HSF homolog, which could explain the discrep-
ancy observed between HSF2 and Drosophila HSF in
binding the resin (Fig. 2). Besides this differential sensitivity
to ionic strength conditions, other components of the
binding buffer did not differentially affect HSF2, except for
MgCl
2
(optimal concentrations: 0 m
M
for HSF2, 1 m
M
for
HSF1), which appeared slightly detrimental to HSF2
binding to DNA (data not shown).
The purified embryonic HSF2 protein displays
a different thermosensitivity than the
i
.
v
.s. factor
I.v.s. HSF1 and HSF2 proteins display very distinct
behaviors. HSF1 protein produced in reticulocyte lysates
is active for DNA binding, provided that the extracts have
first been heated. In contrast, HSF2 protein shows a
constitutive HSE-binding activity but loses this activity
upon heat treatment [3,32]. Therefore, it appeared that the
DNA-binding activity of HSF2 protein was much more
sensitive than that of HSF1 protein, at least when synthes-
ized in vitro.
Using electromobility shift assay (EMSA), we analyzed
the thermosensitivity properties of the purified embryonic
HSF2 in comparison with the i.v.s. factor, produced in
reticulocyte lysates.
I.v.s. or embryonic purified proteins were incubated at
various temperatures before being subjected to EMSA. This
experiment allowed to analyze the sensitivity properties of
soluble HSF2 proteins, by measuring their remaining
capacity to bind their target sequences after exposure to
denaturating temperatures. The remaining ability of the
factors to bind a consensus target was quantified and
plotted as a function of time.
The inactivation ratio of pure embryonic HSF2 protein
was estimated to be about 20% after 20 min at 37 Cand
80% after 20 min at 45 C (Fig. 3). I.v.s. HSF2 protein was
also denatured by incubation at 37 Cor45C(Fig.3).At
high temperatures, the i.v.s. factor appeared to be signifi-
cantly more rapidly inactivated than the embryonic factor.
The inactivation of the i.v.s. protein observed at 37 C
occurred in a limited manner and, unexpectedly, was
preceded by a transient phase of activation. Therefore,
incubation of the i.v.s. HSF2 at a moderate temperature
highly activated its DNA-binding abilities. This result was
uppermost striking as HSF2 appeared to be quite sensitive to
high temperature when synthesized in vitro [3]. Furthermore,
the pure embryonic factor did not behave in the same way.
Thus, HSF2 protein synthesized in the reticulocyte lysates
displayed a specific ability to become further activated
following a short exposure to a moderate temperature. This
seemed not to be characteristic of the factor itself but rather
of the conditions in which it had been produced.
The purified
i
.
v
.s. and embryonic HSF2 proteins exhibit
differences of cooperativity in DNA binding
In order to look for the cooperativity of HSF2 binding to
HSE sequences, we used the same methodology as that
Fig. 2. Effect of ionic (NaCl) strength on Heat-Shock Factors 1 and 2
DNA binding activities. Whole cell extracts from control unshocked
(F9C, corresponding to HSF2) or heat-shocked (F9HS, corresponding
to HSF1) F9 cells were incubated with labeled HSE oligonucleotide
under varying NaCl concentrations. After
PHOSPHORIMAGER
quanti-
fication, data were reported as fractions of the maximal value. Extracts
from heat-shocked cells (F9HS, HSF1) are plotted as circles; extracts
from control cells (F9C, HSF2) are plotted as triangles.
Fig. 1. Purification to homogeneity of HSF2 from F9 tumor extracts.
Elution fractions from the first and second cycle of HSE-affinity col-
umn (as well as HSF2 synthesized in reticulocyte lysates) were run on
SDS/PAGE after 100-fold concentration. (A) Silver staining. The
multiple bands observed above the 70 kDa i.v.s. HSF2 likely corres-
pond to additional proteins present in reticulocyte lysates. The smear
observed above the 70 kDa purified embryonic HSF2 is due to
remaining salts. (B) Western blot analysis using the HSF2 antiserum at
a 1 : 5000 concentration.
FEBS 2002 Determining the optimal binding sequence for HSF2 (Eur. J. Biochem. 269) 2531








![Báo cáo seminar chuyên ngành Công nghệ hóa học và thực phẩm [Mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20250711/hienkelvinzoi@gmail.com/135x160/47051752458701.jpg)






