Crystal structure of a subtilisin-like serine proteinase from
a psychrotrophic Vibrio species reveals structural aspects
of cold adaptation
Jo
´hanna Arno
´rsdo
´ttir
1
, Magnu
´s M. Kristja
´nsson
2
and Ralf Ficner
1
1 Abteilung fu
¨r Molekulare Strukturbiologie, Institut fu
¨r Mikrobiologie und Genetik, Georg-August Universita
¨tGo
¨ttingen, Germany
2 Department of Biochemistry, Science Institute, University of Iceland, Reykjavı
´k, Iceland
Microorganisms inhabit the most diverse environments
on earth. Extremophiles are microorganisms that have
adapted to environmental conditions regarded by
humans as falling outside the normal range in terms of
temperature, pressure, salinity or pH. Extremophiles
have had to develop strategies to deal with environ-
mental stress, mainly by molecular adaptation of their
cell inventory. Of major importance in adapting to
extreme environmental conditions is the optimization
of protein function and stability. Enzymes from
extremophiles are essentially like their mesophilic
counterparts, sharing the same overall fold and
catalysing identical reactions via the same mechanisms,
while having adopted different traits regarding kinetic
and structural properties. Therefore, they provide
excellent tools for examining the molecular basis of
different protein properties, as well as the relation
between structure and function in enzymes. Regarding
temperature, organisms have been isolated from places
with temperatures as high as 113 C [1] and biological
activity has been detected in microbial samples at tem-
peratures as low as )20 C [2]. Thermo- and hyper-
thermophiles face the challenge of keeping their
macromolecules functional under the environmental
Keywords
cold adaptation; crystal structure;
psychrotrophic; subtilase; thermostability
Correspondence
R. Ficner, Abteilung fu
¨r Molekulare
Strukturbiologie, Institut fu
¨r Mikrobiologie
und Genetik, Universita
¨tGo
¨ttingen, Justus-
von-Liebig-Weg11, 37077 Go
¨ttingen,
Germany
Fax: +49 551 391 4082
Tel: +49 551 391 4072
E-mail: rficner@gwdg.de
Database
The coordinates and structure factors for
the final structure of Vibrio proteinase at
1.84 A
˚resolution have been deposited in
the Protein Data Bank under the accession
number 1SH7.
(Received 30 September 2004, revised 26
November 2004, accepted 9 December
2004)
doi:10.1111/j.1742-4658.2005.04523.x
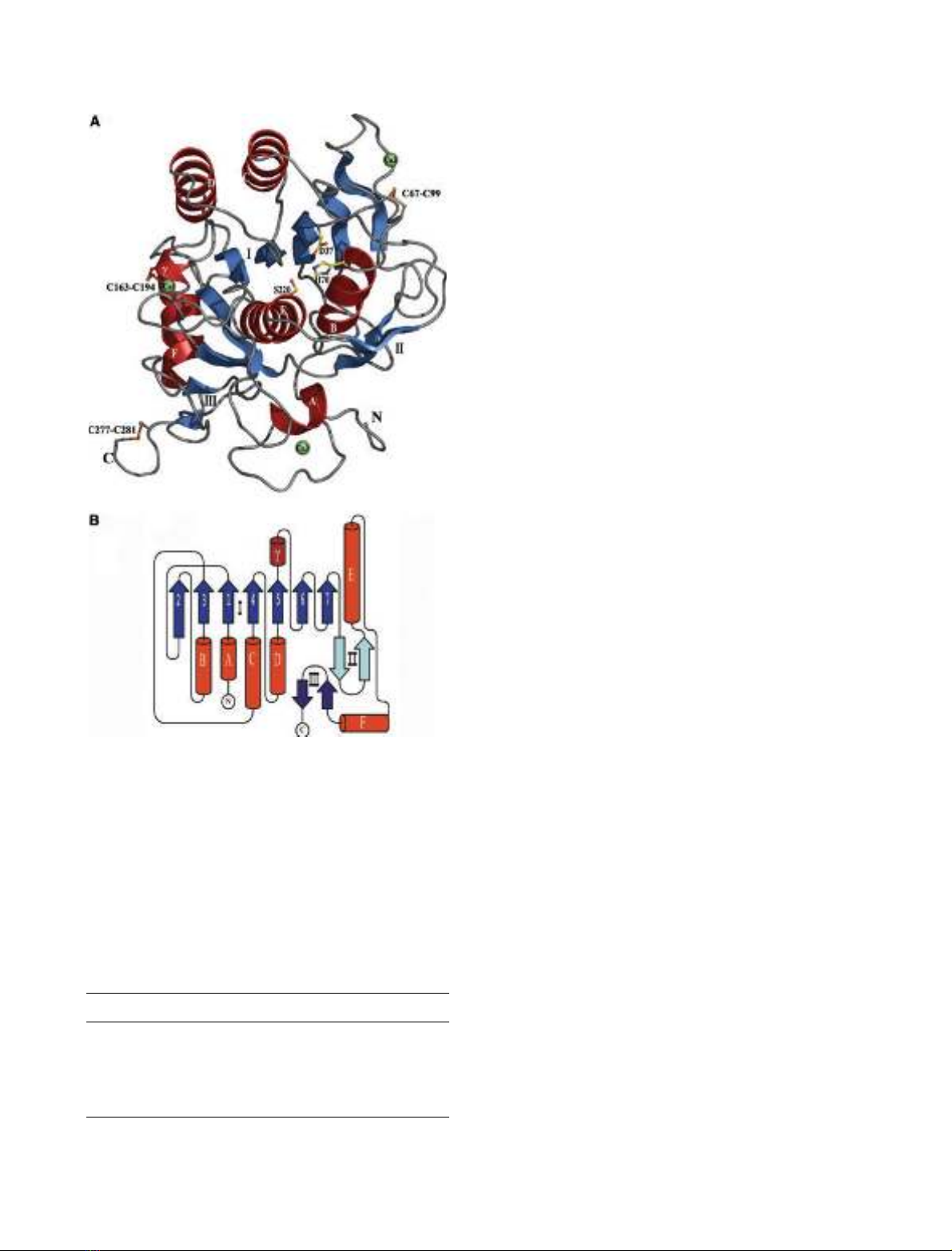
The crystal structure of a subtilisin-like serine proteinase from the psychro-
trophic marine bacterium, Vibrio sp. PA-44, was solved by means of
molecular replacement and refined at 1.84 A
˚. This is the first structure of a
cold-adapted subtilase to be determined and its elucidation facilitates
examination of the molecular principles underlying temperature adaptation
in enzymes. The cold-adapted Vibrio proteinase was compared with known
three-dimensional structures of homologous enzymes of meso- and thermo-
philic origin, proteinase K and thermitase, to which it has high structural
resemblance. The main structural features emerging as plausible determi-
nants of temperature adaptation in the enzymes compared involve the char-
acter of their exposed and buried surfaces, which may be related to
temperature-dependent variation in the physical properties of water. Thus,
the hydrophobic effect is found to play a significant role in the structural
stability of the meso- and thermophile enzymes, whereas the cold-adapted
enzyme has more of its apolar surface exposed. In addition, the cold-adap-
ted Vibrio proteinase is distinguished from the more stable enzymes by its
strong anionic character arising from the high occurrence of uncompen-
sated negatively charged residues at its surface. Interestingly, both the cold-
adapted and thermophile proteinases differ from the mesophile enzyme in
having more extensive hydrogen- and ion pair interactions in their struc-
tures; this supports suggestions of a dual role of electrostatic interactions
in the adaptation of enzymes to both high and low temperatures. The
Vibrio proteinase has three calcium ions associated with its structure, one
of which is in a calcium-binding site not described in other subtilases.
832 FEBS Journal 272 (2005) 832–845 ª2005 FEBS