Crystal structures of isomaltase from
Saccharomyces cerevisiae and in complex with its
competitive inhibitor maltose
Keizo Yamamoto
1
, Hideo Miyake
2
, Masami Kusunoki
3
and Shigeyoshi Osaki
1
1 School of Medicine, Nara Medical University, Japan
2 Graduate School of Bioresources, Mie University, Japan
3 Faculty of Engineering, University of Yamanashi, Japan
Introduction
Oligo-1,6-glucosidase (EC 3.2.1.10; oligosaccharide
oligo-1,6-glucosidase) hydrolyzes the a-1,6-glucosidic
linkage of isomalto-oligosaccharides and dextran [1,2].
However, unlike a-1,4-glucosidases (EC 3.2.1.20), oligo-
1,6-glucosidase fails to hydrolyze the a-1,4-glucosidic
bonds of maltosaccharides [2–8].
On the basis of amino acid sequence similarities, oligo-1,
6-glucosidase has been classified as a member of the
retaining glycoside hydrolase (GH) family 13, also
called the a-amylase family. GH family 13 includes
a-amylases, a-glucosidases, cyclodextrin glucantransfe-
rases, pullulanases, isoamylases, branching enzymes,
and neopullulanases [9,10]. The members of GH
family 13 share little amino acid sequence similarity.
However, they contain four highly conserved regions
(regions I–IV), and three catalytic acidic residues
Keywords
crystal structure; glycoside hydrolase
family 13; isomaltase; substrate binding;
substrate specificity
Correspondence
K. Yamamoto, School of Medicine, Nara
Medical University, 840 Shijo, Kashihara,
Nara 634-8521, Japan
Fax: +81 744 29 8810
Tel: +81 744 29 8810
E-mail: kama@naramed-u.ac.jp
Database
Structural data are available in the Protein
Data Bank under the accession numbers
3AJ7 and 3A4A
(Received 2 June 2010, revised 27 July
2010, accepted 5 August 2010)
doi:10.1111/j.1742-4658.2010.07810.x
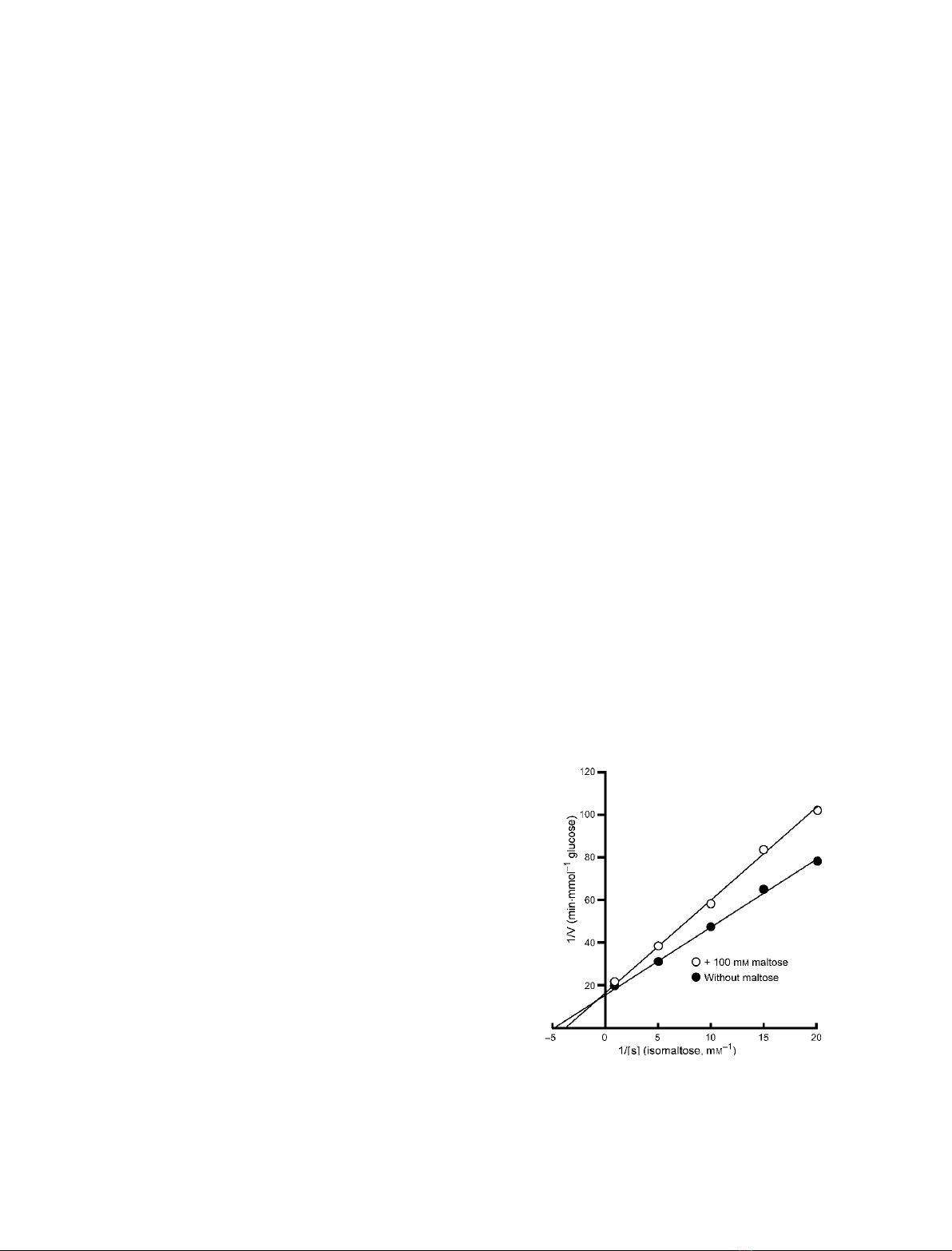
The structures of isomaltase from Saccharomyces cerevisiae and in complex
with maltose were determined at resolutions of 1.30 and 1.60 A
˚, respec-
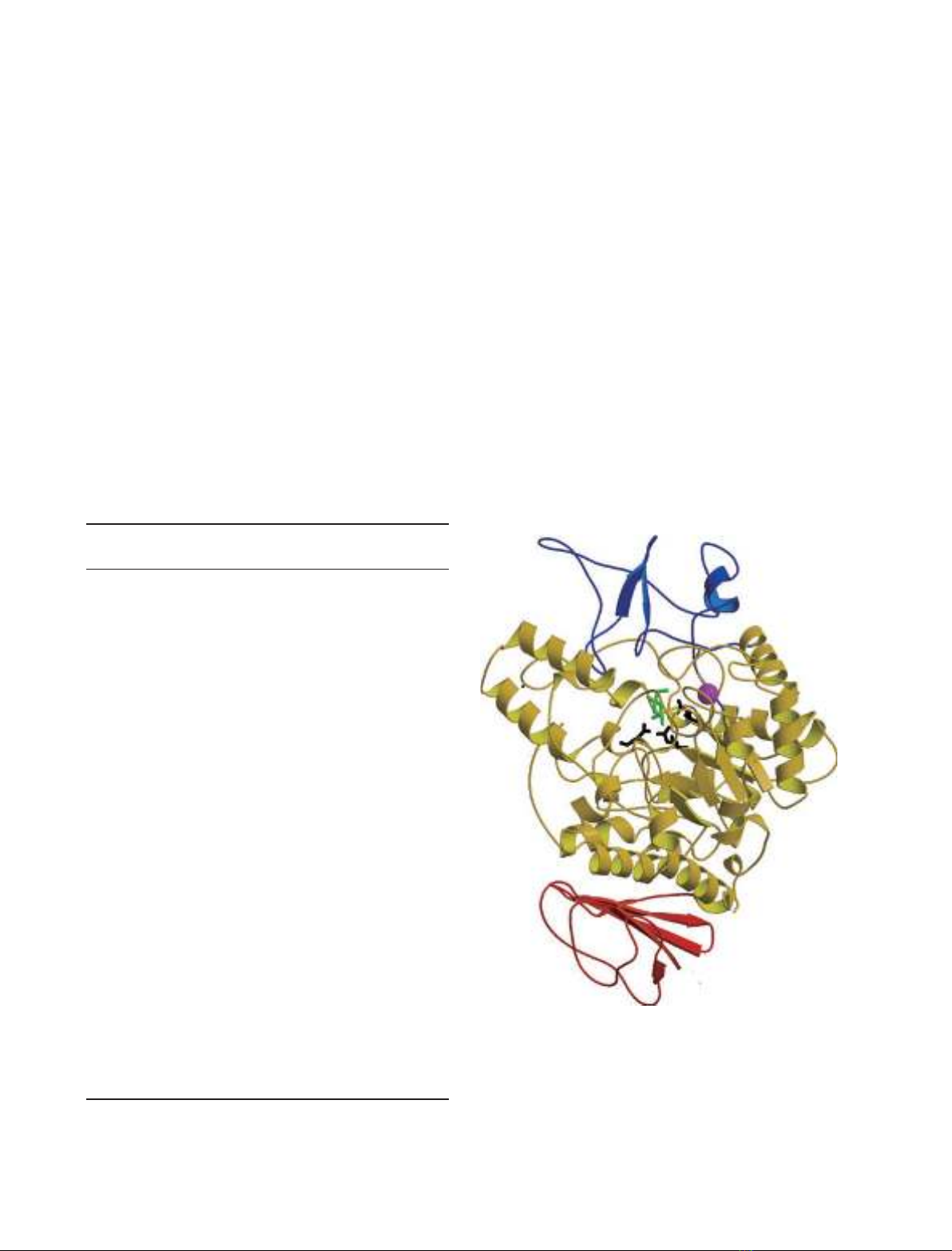
tively. Isomaltase contains three domains, namely, A, B, and C. Domain A
consists of the (b⁄a)
8
-barrel common to glycoside hydrolase family 13.
However, the folding of domain C is rarely seen in other glycoside hydro-
lase family 13 enzymes. An electron density corresponding to a nonreduc-
ing end glucose residue was observed in the active site of isomaltase in
complex with maltose; however, only incomplete density was observed for
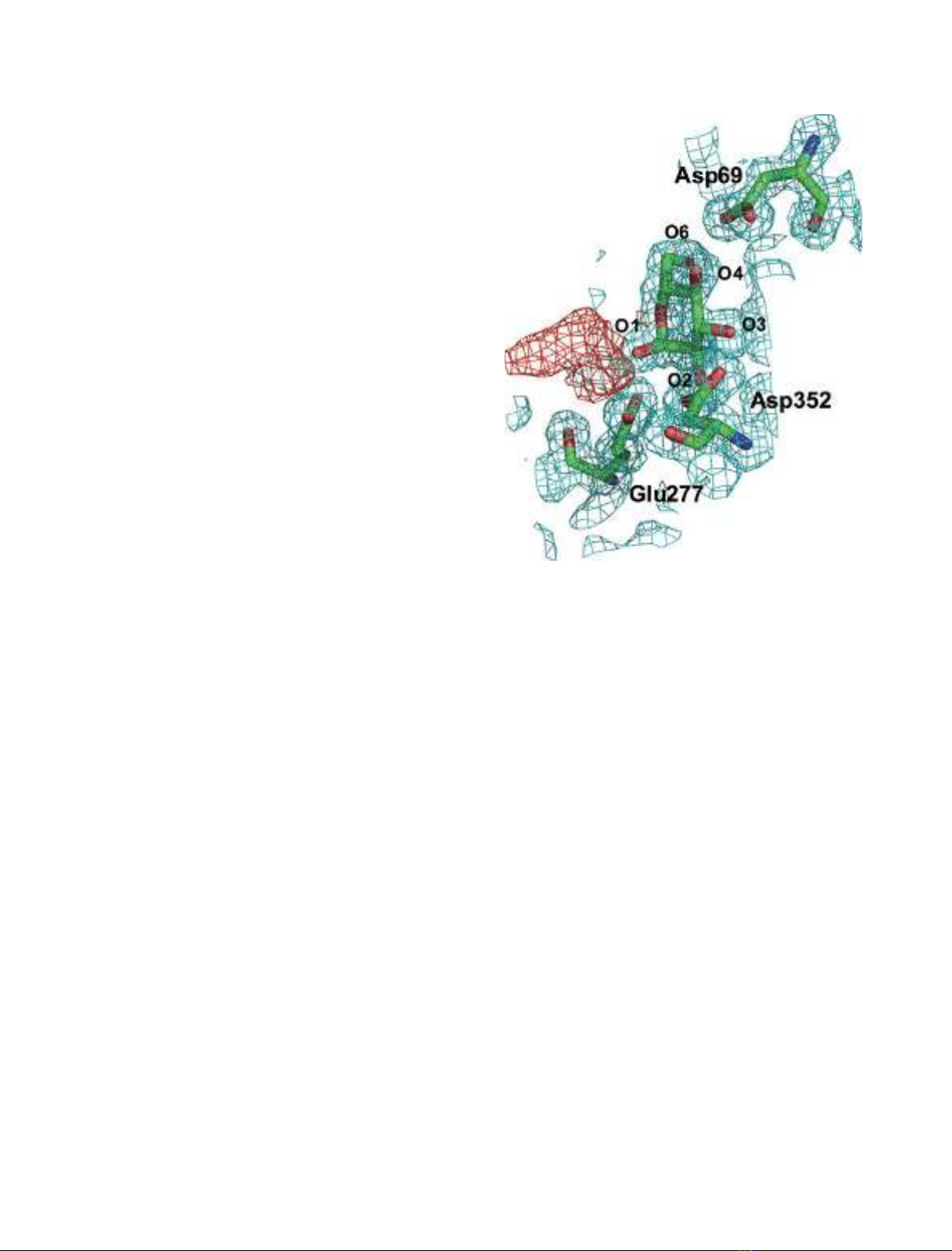
the reducing end. The active site pocket contains two water chains. One
water chain is a water path from the bottom of the pocket to the surface
of the protein, and may act as a water drain during substrate binding. The
other water chain, which consists of six water molecules, is located near
the catalytic residues Glu277 and Asp352. These water molecules may act
as a reservoir that provides water for subsequent hydrolytic events. The
best substrate for oligo-1,6-glucosidase is isomaltotriose; other, longer-
chain, oligosaccharides are also good substrates. However, isomaltase
shows the highest activity towards isomaltose and very little activity
towards longer oligosaccharides. This is because the entrance to the active
site pocket of isomaltose is severely narrowed by Tyr158, His280, and loop
310–315, and because the isomaltase pocket is shallower than that of other
oligo-1,6-glucosidases. These features of the isomaltase active site pocket
prevent isomalto-oligosaccharides from binding to the active site
effectively.
Abbreviations
DGase, dextran glucosidase; GH, glycoside hydrolase; O16G, oligo-1,6-glucosidase from Bacillus cereus;a-MG, methyl a-D-glucopyroside.
FEBS Journal 277 (2010) 4205–4214 ª2010 The Authors Journal compilation ª2010 FEBS 4205