
The Fe-only nitrogenase and the Mo nitrogenase
from
Rhodobacter capsulatus
A comparative study on the redox properties of the metal clusters
present in the dinitrogenase components
Stefan Siemann*, Klaus Schneider, Melanie Dro¨ ttboom† and Achim Mu¨ ller
Lehrstuhl fu
¨r Anorganische Chemie I, Fakulta
¨tfu
¨r Chemie der Universita
¨t Bielefeld, Bielefeld, Germany
The dinitrogenase component proteins of the conventional
Mo nitrogenase (MoFe protein) and of the alternative
Fe-only nitrogenase (FeFe protein) were both isolated and
purified from Rhodobacter capsulatus, redox-titrated
according to the same procedures and subjected to an EPR
spectroscopic comparison. In the course of an oxidative
titration of the MoFe protein (Rc1
Mo
) three significant
S¼1/2 EPR signals deriving from oxidized states of the
P-cluster were detected: (1) a rhombic signal (g¼2.07, 1.96
and 1.83), which showed a bell-shaped redox curve with
midpoint potentials (E
m
)of)195 mV (appearance) and
)30 mV (disappearance), (2) an axial signal (g
||
¼2.00,
g
^
¼1.90) with almost identical redox properties and (3) a
second rhombic signal (g¼2.03, 2.00, 1.90) at higher redox
potentials (> 100 mV). While the Ôlow-potentialÕrhombic
signal and the axial signal have been both attributed to the
one-electron-oxidized P-cluster (P
1+
) present in two con-
formationally different proteins, the Ôhigh-potentialÕrhombic
signal has been suggested rather to derive from the P
3+
state.
Upon oxidation, the FeFe protein (Rc1
Fe
) exibited three
significant S¼1/2 EPR signals as well. However, the Rc1
Fe
signals strongly deviated from the MoFe protein signals,
suggesting that they cannot simply be assigned to different
P-cluster states. (a) The most prominent feature is an
unusually broad signal at g¼2.27 and 2.06, which proved
to be fully reversible and to correlate with catalytic activity.
The cluster giving rise to this signal appears to be involved in
the transfer of two electrons. The midpoint potentials
determined were: )80 mV (appearance) and 70 mV (dis-
appearance). (b) Under weakly acidic conditions (pH 6.4) a
slightly altered EPR signal occurred. It was characterized by
a shift of the gvalues to 2.22 and 2.05 and by the appearance
of an additional negative absorption-shaped peak at
g¼1.86. (c) A very narrow rhombic EPR signal at
g¼2.00, 1.98 and 1.96 appeared at positive redox potentials
(E
m
¼80 mV, intensity maximum at 160 mV). Another
novel S¼1/2 signal at g¼1.96, 1.92 and 1.77 was observed
on further, enzymatic reduction of the dithionite-reduced
state of Rc1
Fe
with the dinitrogenase reductase component
(Rc2
Fe
) of the same enzyme system (turnover conditions in
the presence of N
2
and ATP). When the Rc1
Mo
protein was
treated analogously, neither this Ôturnover signalÕnor any
other S¼1/2 signal were detectable. All Rc1
Fe
-specific EPR
signals detected are discussed and tentatively assigned with
special consideration of the reference spectra obtained from
Rc1
Mo
preparations.
Keywords: Fe nitrogenase; FeFe cofactor; FeMo cofactor;
P-cluster; EPR spectroscopy.
Four types of nitrogenase systems have been demonstrated
to exist in bacteria and archea so far. They have been clearly
shown to be genetically as well as biochemically distinct.
The first genetic nitrogen fixation (nif ) system discovered is
responsible for encoding the conventional molybdenum
(Mo)-containing nitrogenase. Two nitrogenase systems are
closely related to the Mo nitrogenase, but Mo-independent.
One is the vanadium (V)-dependent nitrogen fixation (vnf )
system encoding a nitrogenase which contains V instead of
Mo in the cofactor (vanadium nitrogenase) [1–4], whereas
the other, represented by the alternative nitrogen fixation
(anf ) gene system, encodes a nitrogenase containing neither
Mo, V nor any other heterometal atom [4–9], and has
therefore been designated as the Fe nitrogenase or Fe-only
nitrogenase. Recently, a heterotrimeric and completely
nif/vnf/anf-independent nitrogenase system has been repor-
tedtooccurinStreptomyces thermoautotrophicus,inwhich
electrons for N
2
reduction are derived from superoxide
oxidation coupled to CO oxidation [10].
Correspondence to A. Mu
¨ller, Lehrstuhl fu
¨rAnorganischeChemieI,
Fakulta
¨tfu
¨r Chemie, Universita
¨t Bielefeld, Postfach 100131, 33501
Bielefeld, Germany. Fax: + 49 521 1066003,
Tel.: + 49 521 1066153, E-mail: a.mueller@uni-bielefeld.de
Abbreviations:nif, nitrogen fixation; vnf, vanadium dependent nitro-
gen fixation; anf, alternative nitrogen fixation; FeMoco, iron–molyb-
denum cofactor; FeFeco, iron–iron cofactor; Rc1
Mo
, MoFe protein of
R. capsulatus;Rc1
Fe
,FeFeproteinofR. capsulatus;Rc2
Mo
,Fepro-
tein of the Mo nitrogenase of R. capsulatus;Rc2
Fe
, Fe protein of the
Fe-only nitrogenase of R. capsulatus; EXAFS, extended X-ray
absorption fine structure.
Enzyme: nitrogenase (EC 1.18.6.1).
*Present address: Department of Chemistry, University of Waterloo,
Waterloo, Ontario, Canada.
Present address: Transferstelle Umweltbiotechnology,
Ruhr-Universita
¨t Bochum, 44780 Bochum, Germany.
(Received 19 September 2001, revised 28 December 2001, accepted
22 January 2002)
Eur. J. Biochem. 269, 1650–1661 (2002) ÓFEBS 2002

The characteristics of Mo, V and Fe nitrogenases have
been reviewed recently by Eady [3] and Smith [4]. All three
nitrogenase systems consist of two-component proteins, the
dinitrogenase component (MoFe protein, VFe protein,
FeFe protein) and the dinitrogenase-reductase component
(also termed Fe protein with respect to all three types of
nitrogenases). While the MoFe protein consists of four
subunits forming an a
2
b
2
tetramer, the dinitrogenase
proteins of the Mo-independent, alternative nitrogenases,
contain an additional small 13–15 kDa subunit to form an
a
2
b
2
d
2
hexameric structure.
The dinitrogenase component of nitrogenases contains
two types of unique metal clusters, the so-called M-cluster
(FeMo cofactor, FeV cofactor, FeFe cofactor), which
represents the site of substrate reduction [11], and the
P-cluster, whose function is likely to transfer electrons as well
as protons to the cofactor [12]. Based on X-ray crystal
structure analysis of MoFe proteins, the structures of the
FeMo cofactor (Fe
7
MoS
9
/homocitrate) and the P-cluster
(Fe
8
S
7
) have been elucidated [12,13], the specific site(s) of
substrate binding and reduction within the cofactor, how-
ever, still remain a matter of controversial discussion [14–17].
So far, only three Fe-only nitrogenases have been
genetically (as anf systems) as well as biochemically
identified and characterized. These are the enzymes of
Azotobacter vinelandii [5,6], Rhodospirillum rubrum [9] and
Rhodobacter capsulatus [8,18,19], the heterometal-free
N
2
-fixation system from the latter organism being the most
intensively studied.
During the early years of Fe nitrogenase research, doubts
were widespread as to whether an Fe-only nitrogenase can
be isolated as an intact, functioning enzyme. These doubts
primarily arose due to the fact that preparations of the type
of anf-dependent nitrogenase were, regardless of their origin,
generally characterized by either extremely low catalytic
activity [5,6,9,18] or the wrong cofactor (namely the FeMo
cofactor) incorporated into the alternative dinitrogenase
component [6,19]. However, a comprehensive characteriza-
tion of the Fe-only nitrogenase isolated from R. capsulatus,
which included a detailed comparison with the Mo-contain-
ing nitrogenase from the same organism, showed that: (a) the
Fe nitrogenase components can indeed be isolated and
purified as intact and catalytically active proteins, and
(b) that the FeFe protein definitely does not contain an
iron–molybdenum cofactor (FeMoco), but a clearly well-
functioning Fe-only cofactor [8]. Relatively high specific
activities have been reported for N
2
reduction (350 nmol of
NH
3
formed per min per mg protein), acetylene reduction
as well as very high activities (1300 nmol H
2
Æmin
)1
Æmg
)1
in
an N
2
atmosphere) for the evolution of molecular hydrogen
[8]. It is interesting to note that, particularly in the
simultaneous presence of a second substrate (N
2
or C
2
H
2
in addition to H
+
), the H
2
production rates were distinctly
higher than the respective activities of the Mo nitrogenase
(sixfold). Samples of such highly active FeFe protein
preparations contained 26 ± 4 Fe atoms per protein
molecule, but neither molybdenum nor vanadium [8].
A recent
57
Fe-Mo
¨ssbauer-/Fe-EXAFS study on the FeFe
protein from R. capsulatus provided strong evidence that:
(a) the FeFe cofactor is diamagnetic in the Na
2
S
2
O
4
-
reduced state containing 4Fe
II
and 4Fe
III
centers, and (b) the
main structural feature of the FeMoco, the central trigonal
prismatic arrangement of Fe atoms, is also present in the
FeFe cofactor, thus indicating a structural homology
between both cofactor types [20,21].
A definite identification of the Fe-only cofactor by EPR is
still lacking. Nevertheless, based on the results of preceding
investigations [8], the FeFe protein exhibited several inter-
esting and, with respect to the MoFe protein, deviating EPR
spectroscopic properties. (a) Highly active FeFe protein
samples (reduced with Na
2
S
2
O
4
) neither showed a FeMoco-
typical S¼3/2 EPR signal nor any other signal indicative
of a S¼3/2 spin system. Instead they were, in agreement
with the analysis of Mo
¨ssbauer spectra [21], EPR silent.
(b) A novel S¼1/2 signal (g¼1.96, 1.92, 1.77) appeared
on dinitrogenase reductase-mediated reduction of the FeFe
protein (turnover conditions). (c) Two further significant
EPR signals were observed when the FeFe protein was
partially oxidized with K
3
[Fe(CN)
6
] or thionine: an unusu-
ally broad signal centered at g¼2.27 and 2.06 and a very
narrow rhombic signal at g¼2.00, 1.98 and 1.96.
A conclusive assignment of these novel EPR signals to
either the cofactor or the P-cluster has proven elusive due to
the fact that both of these metal clusters present in the
Fe-only nitrogenase are diamagnetic in the dithionite-
reduced state, but probably become EPR-active upon
oxidation.
In the present work we focused on the identification or
tentative assignment of the most significant EPR signals
detected with FeFe protein samples, by pursuing the
following approach: the FeFe and the MoFe proteins were
isolated from the same organism, samples were prepared
according to the same procedures and subsequently char-
acterized and compared by EPR spectroscopy, particularly
with respect to their redox properties.
MATERIALS AND METHODS
Bacterial strains
The organisms used were the R. capsulatus wild-type strain
B10S and the Mo-resistant double mutant with a nifHDK
deletion as well as an additional deletion in the modABCD
region [19,22]. The products of the latter genes are involved
in high-affinity molybdenum transport [22].
Growth medium and culture conditions
The growth medium and culture conditions applied were as
described previously [8].
Purification of nitrogenase proteins
Preparation of cell-free extracts (cell disruption by lysozyme
followed by ultracentrifugation) were performed as des-
cribed by Schneider et al.[8].Inviewofthedifficultyin
separating the dinitrogenase (Rc1
Mo
) and dinitrogenase
reductase component (Rc2
Mo
) of the Mo nitrogenase
from R. capsulatus by DEAE chromatography, we used
Q-Sepharose (from Pharmacia), a stronger and more
effective anion exchanger, for the purification of both the
Fe-only and the Mo nitrogenase components. The column
(internal diameter: 2.5 cm) containing approximately
60 mL gel, was cooled to 8 °C with a cryostat and
equilibrated with Ar-gassed Tris buffer (50 m
M
,pH7.8)
containing NaCl (150 m
M
) and sodium dithionite (2 m
M
).
ÓFEBS 2002 Redox properties of the FeFe protein (Eur. J. Biochem. 269) 1651

The cell-free extract was loaded onto the Q-Sepharose
column, followed by the stepwise elution with approxi-
mately 50–60 mL of NaCl solutions (in equilibration buffer)
of increasing concentrations (200/250/300/350/400 m
M
in
the case of the Mo nitrogenase and 200/250/280/310/340/
400 m
M
in the case of the Fe nitrogenase). The Rc1
Mo
component was eluted with 300 m
M
NaCl, whereas Rc2
Mo
was recovered in the 350 m
M
NaCl fraction. In the case of
theFenitrogenasetheRc2
Fe
component was eluted with
280 m
M
NaCl prior to the recovery of Rc1
Fe
with 330 m
M
NaCl. All nitrogenase component proteins were concentra-
ted to approximately 8 mL by anaerobic ultrafiltration in a
50-mL chamber equipped with a PM30 Amicon membrane,
and subsequently further concentrated to a final volume of
1 mL in a B15 Amicon chamber. Both dinitrogenase
components, which were of relevance for the present
comparative EPR study (Rc1
Mo
,Rc1
Fe
), were, based on
SDS/PAGE analysis, 90–95% pure.
The protocol previously employed to purify the MoFe
protein (DEAE chromatography, Sephadex G-150 gel-
filtration) [8] led to a homogeneous preparation with
significantly lower protein yield. Because the EPR spectra
of samples obtained from both the Q-Sepharose and the
DEAE/gel-filtration procedures were indistinguishable, we
preferred the use of the rapid and high-yield one-column
method (Q-Sepharose) also for the purification of the MoFe
protein in the present study.
Determination of nitrogenase activity
and protein content
For the determination of nitrogenase activity the routine
assay (C
2
H
2
reduction) was employed [8]. Protein was
determined according to Beisenherz et al.[23].
Metal and acid-labile sulfide determinations
The quantitative determination of Fe and Mo was achieved
by inductively coupled plasma mass spectrometry as repor-
ted previously [24]. Fe was additionally determined by the
bathophenanthrolin method [2]. Acid-labile sulfide analysis
was performed according to Chen & Mortenson [25].
Redox titrations
Redox titrations were performed in a modified redox
titration cell similar to that described by Dutton [26]. The
redox potential was measured with a combined platinum-
Ag/AgCl electrode (PT 4800-M5-S7/80; Mettler Toledo,
Steinbach, Germany) and the achieved potentials were
quoted relative to the standard hydrogen electrode. The
method involved titrating the protein in Hepes buffer
(50 m
M
,pH7.4)at25°C in the presence of the following
mediators (each at 43 l
M
): 2,6-dichlorophenolindophenol,
phenazine methosulfate, thionine, methylene blue, indigo
trisulfonate, indigo carmine, resorufin, anthraquinone-
2-sulfonate, safranin O, benzyl viologen, methyl viologen.
Prior to the redox titration, the protein sample was
subjected to buffer exchange by gel filtration on Sephadex
G25 equilibrated with 50 m
M
Hepes (pH 7.4) containing
1m
M
Na
2
S
2
O
4
(sodium dithionite). It is pertinent to note
that the reducing agent was not entirely removed from FeFe
protein preparations in view of the lability of the protein
even in the presence of only trace amounts of oxygen [8].
For the sake of direct comparison, MoFe protein samples
were treated under analogous conditions.
The final sample solution (3 mL) containing 12–14 mg of
protein per mL was adjusted to different redox potentials by
the stepwise addition (0.5 lL) of K
3
[Fe(CN)
6
] (ferricyanide)
as oxidant and Na
2
S
2
O
4
as reductant. After equilibration,
which was usually achieved after 1–2 min, 170-lLsamples
were withdrawn from the solution with a gas-tight syringe,
placed in an EPR tube and immediately frozen in liquid N
2
for EPR spectroscopic measurements.
EPR measurements
EPR (X band) spectra were recorded on a Bruker ECS 106
spectrometer equipped with an ECS 041 MR Bruker
microwave bridge and an Oxford Instruments EPR 900
helium flow cryostat. All spectra were recorded at a
microwave frequency of 9.44 GHz and a field modulation
of 1.0 mT at 100 kHz. Spin quantification was performed
using 10 m
M
CuSO
4
/10 m
M
HCl/2
M
NaClO
4
as an
external standard for integration.
RESULTS
EPR signals from oxidized states of the MoFe protein
In recent years EPR spectroscopic properties have been
reported for several MoFe proteins, mainly focusing on
P-cluster-type signals [27–31]. Based on the notion,
however, that, dependent on the origin, the purification
procedure and the sample quality (specific activity),
considerable differences within one class of enzyme may
occur, we did not rely on literature data, but attempted
the direct experimental comparison of the MoFe and the
FeFe protein. We therefore isolated and prepared both
proteins not only from the same organism (R. capsulatus)
but also under the same conditions (lysozymatic cell
disruption, Q-Sepharose chromatography, EPR sample
preparation). For EPR experiments, protein samples were
used which displayed approximately maximal specific
activities, i.e. 250 U (nmol acetylene reducedÆmin
)1
)
per mg of FeFe protein and 1000–1200 UÆmg
)1
of MoFe
protein (compare [8]).
In the course of these studies two experimental routes to
obtain different redox states of the dinitrogenase protein
were pursued: (a) a rough, stepwise oxidation with
K
3
[Fe(CN)
6
] and (b) a redox titration, adjusting the protein
solution to defined potentials in the presence of redox
mediators.
Stepwise oxidation of the MoFe protein. In its Na
2
S
2
O
4
-
reduced state the R. capsulatus MoFe protein (Rc1
Mo
) only
exhibited the characteristic S¼3/2 EPR signal at g¼4.29,
3.67 and 2.01, arising from the cofactor (compare Fig. 6B,
which presents a signal-comparison of the dithionite-
reduced and the turnover state of Rc1
Mo
). In the same
redox state the P-cluster was EPR-silent (P
N
state). Upon
oxidation two significant types of P-cluster signals appeared.
When samples (pH 7.4), reduced with 1 m
M
dithionite,
were supplemented with successively increasing amounts of
K
3
[Fe(CN)
6
], a rhombic S¼1/2 EPR signal at g¼2.07,
1.96 and 1.83 appeared (Fig. 1, spectrum 1). This signal was
1652 S. Siemann et al. (Eur. J. Biochem. 269)ÓFEBS 2002
most prominent with 2 m
M
K
3
[Fe(CN)
6
] and decreased
again above this concentration. With respect to its shape
and position of the gvalues, this signal appears to
correspond to the S¼1/2 signal that has been reported
for the partially oxidized MoFe proteins from Klebsiella
pneumoniae (Kp1) and A. vinelandii (Av1
Mo
) [28,30,32].
This type of signal has been interpreted to arise from the 1e
–
oxidized P-cluster (P
1+
)[28].
After the occurrence of an almost EPR-silent intermedi-
ate redox state (spectrum not shown), a second rhombic, but
much more narrow EPR signal at g¼2.03, 2.00 and 1.90
appeared upon further oxidation (Fig. 1, spectrum 2). This
signal reached an intensity maximum with 4 m
M
K
3
[Fe(CN)
6
] and remained unchanged with higher oxidant
concentrations. This result suggests that the cluster giving
rise to this signal cannot be oxidized further under the
conditions applied. In studies with Av1
Mo
a similar signal,
although much broader and shifted to distinctly higher
fields (g¼1.97, 1.88, 1.68), has been observed and attrib-
uted to the P-cluster in its 3e
–
oxidized state [27].
Equilibrium-mediated redox titration of the MoFe pro-
tein. The EPR spectroscopic investigation of Rc1
Mo
sam-
ples (in 50 m
M
Hepes buffer, pH 7.4), which were subjected
to a redox titration in the presence of mediators (see
Materials and methods), yielded in parts agreeing, in other
parts, however, somewhat differing spectral data.
In accordance with studies on MoFe proteins from other
organisms (e.g [27]), a midpoint potential (E
m
)of)50 mV
was determined for the S¼3/2 FeMoco signal of Rc1
Mo
(Fig. 2). Above +100 mV the FeMoco signal disappeared
completely.
The EPR signal originating from the 1e
–
oxidized
P-cluster with the central gvalue at 1.96 (in the following
designated as ÔP
1+
signalÕ) appeared at )250 mV, reached
an intensity maximum at )120 mV and decreased again
with increasing potentials. The bell-shaped redox curve of
the P
1+
signal thus confirms the involvement of the
P-cluster in the transfer of at least two electrons (compare
[27,30]). The midpoint potentials determined were:
)195 mV (E
m
for appearance of the signal representing
the P
N/1+
transition) and )30 mV (E
m
for disappearance;
P
1+/2+
transition).
In contrast to the pronounced pH dependence of the P
1+
signal caused by the partially oxidized Av1
Mo
protein [30]
(see Discussion), the Rc1
Mo
-induced P
1+
signal was not
significantly influenced by the pH value. The intensity was
almost identical at pH 6.4 and 7.4 and was still 60% (with
respect to peak height) at pH 8.4. It was, however, a surprise
that, in the course of the redox titration and pH dependence
studies, a new axial S¼1/2 signal in the g¼2region
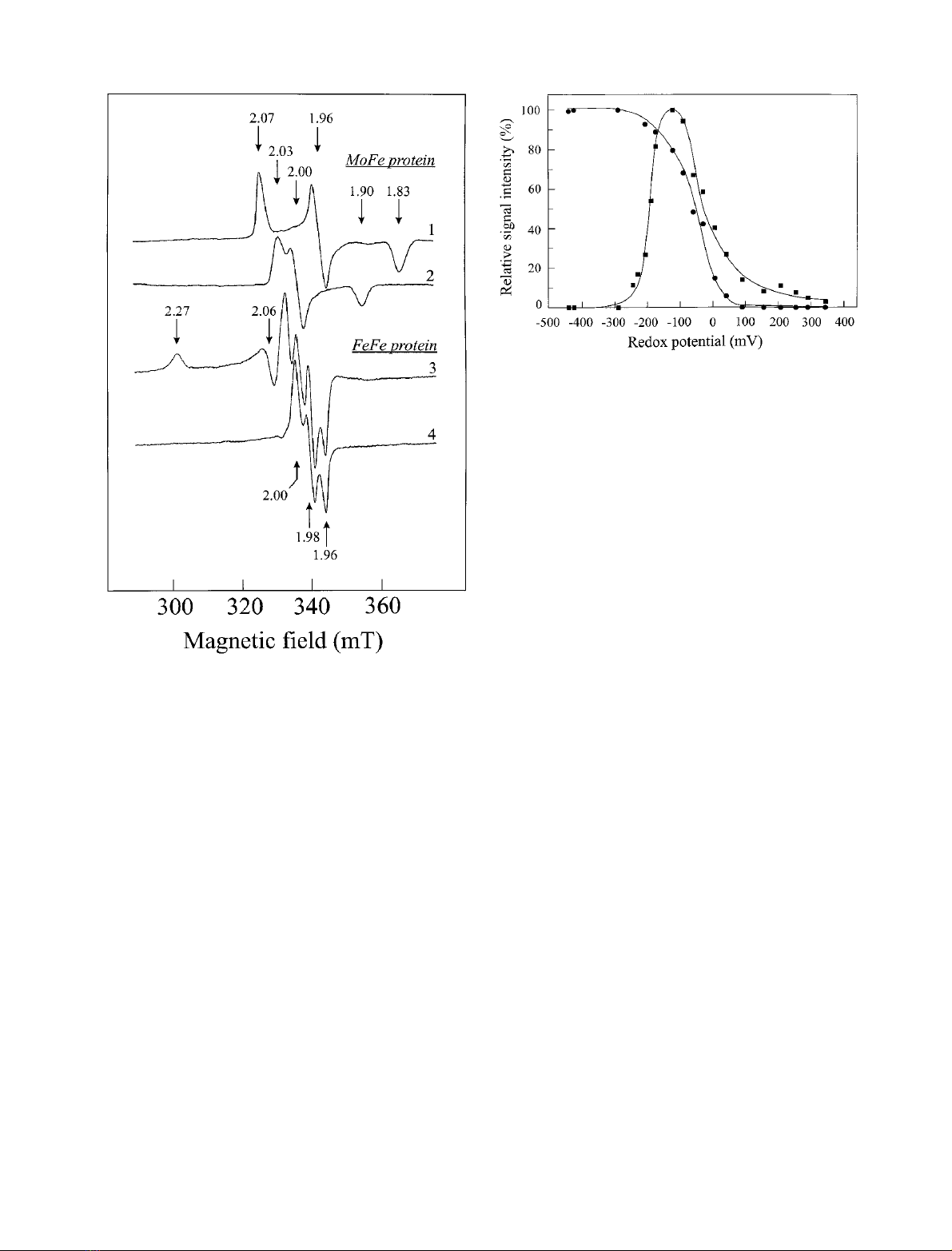
Fig. 1. P-cluster EPR signals of the MoFe protein compared to the EPR
signals detected with the oxidized FeFe protein. TheMoFeprotein
sample contained 21 mg protein per mL, 1.9 (± 0.2) Mo atoms and
27 (± 3) Fe atoms per molecule; the FeFe protein sample contained
18 mg protein per mL and 29 (± 3) Fe atoms per molecule. Both
samples were prepared in 50 m
M
Tris (pH 7.4) containing 1 m
M
Na
2
S
2
O
4
. Spectrum 1, MoFe protein, oxidation with 2 m
M
K
3
[Fe(CN)
6
], measured at 16 K; spectrum 2, MoFe protein, oxidation
with 4 m
M
K
3
[Fe(CN)
6
], measured at 16 K; spectrum 3, FeFe protein,
oxidation with 2.5 m
M
K
3
[Fe(CN)
6
], measured at 10 K; spectrum 4,
FeFe protein, oxidation with 2.5 m
M
K
3
[Fe(CN)
6
], measured at 23 K.
All spectra were recorded at 100 mW.
Fig. 2. Redox titration of the cofactor and P-cluster EPR signals deri-
ving from the MoFe protein of R. capsulatus.The redox titration was
performed as described in Materials and methods. The sample con-
tained 12 mg MoFe protein per mL. (d) Redox titration curve of the
FeMo cofactor signal. For the determination of relative signal intensity
the resonance at g¼3.67 was used. Spectra were measured at 4 K and
20 mW. (j) Redox titration curve of the rhombic P-cluster (P
1+
state)
signal. Intensity determination was performed using the g¼1.96
resonance. Spectra were recorded at 16 K and 100 mW.
ÓFEBS 2002 Redox properties of the FeFe protein (Eur. J. Biochem. 269) 1653
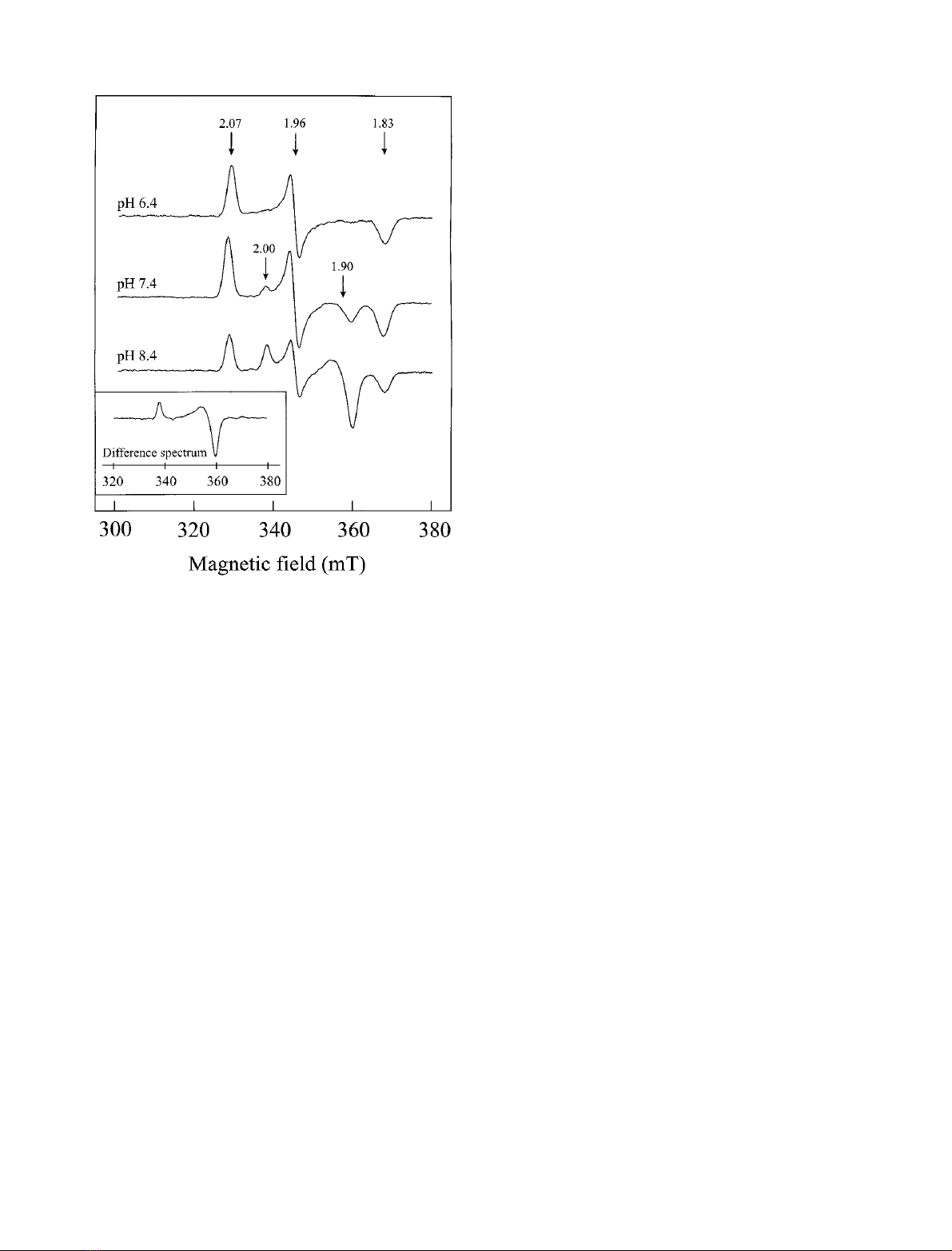
(g
||
¼2.00, g
^
¼1.90) was detected, which showed a
distinctly stronger but, referred to the P
1+
signal of Av1
Mo
,
opposite pH dependence (Fig. 3). The intensity of this signal
was maximal at pH 8.4, with no significant change up to
pH 9.0. At pH 7.4 the signal intensity accounted for
approximately 40% and at pH < 6.5 the signal was absent.
The profile of the entire signal, without interference of the
rhombic signal, was obtained by subtracting the pH 6.4-
spectrum from the pH 8.4-spectrum (see the inset of Fig. 3).
The axial signal and the rhombic P
1+
signal differed
significantly with respect to temperature and microwave-
power dependency. The P
1+
signal was most pronounced
around 18 K, the axial signal around 13 K. While the P
1+
signal appeared to be slightly power saturated already above
25 mW, the axial signal remained unsaturated even at
200 mW. However, both signals behaved similarly with
respect to their dependence on the redox potential. This
observation indicates that the axial signal might arise from
the P
1+
cluster as well, possibly in a slightly modified
environment (protein conformation). It is pertinent to note
that this axial signal is also detectable in the spectrum
obtained after partial oxidation with K
3
[Fe(CN)
6
] without
mediators (at pH 7.4), although with much lower intensity
(data not shown).
The rhombic signal at g¼2.03, 2.00 and 1.90, which
appeared prominently after oxidation with K
3
[Fe(CN)
6
]
(> 4 m
M
) and was proposed to represent the P
3+
state
(Fig. 1, spectrum 4), was only noticeable as a very weak
signal during redox titration (at potentials > 100 mV).
Even excessive amounts of K
3
[Fe(CN)
6
] did not cause a
significant increase in signal intensity.
It is interesting to note that S¼5/2 signals, observed in
the case of Av1
Mo
and attributed to the P
1+
state [28], as
well as S¼7/2 signals (P
3+
state) [27] both simultaneously
present with S¼1/2 signals (forming so-called spin
mixtures), were not detected in the case of the Rhodobacter
enzyme.
At potentials > 0 mV an additional weak signal near
g¼12 was detected (spectrum not shown). In the case of
Av1
Mo
this low field signal has been attributed to the
2e
–
-oxidized P-cluster (S¼3) [27,30]. An exact determin-
ation of the midpoint potential was, however, not possible
due to the low intensity of this signal (integer spin system)
under standard EPR conditions (perpendicular mode).
The two characteristic
S
¼1/2 signals of the partially
oxidized FeFe protein
Stepwise oxidation of the FeFe protein. The protein
preparations used in this study contained 29 (± 3) Fe and
31 (± 4) acid-labile sulfur atoms. The high Fe/S content
indicates that these FeFe protein (Rc1
Fe
) preparations were
virtually devoid of any significant amounts of inactive
(oxidatively damaged clusters) or incompletely assembled
(vacant cofactor sites) enzyme. It is interesting to note that
in the case of dithionite-reduced VFe proteins [3,33] and
also in some instances with MoFe proteins [27,34] both such
protein forms gave rise to S¼1/2 signals. In sharp
contrast, the Rc1
Fe
protein is, in agreement with the
preceding report [8], apparently EPR silent in the presence
of excess dithionite. Neither an S¼3/2 nor a significant
S¼1/2 signal in the g¼2 region (< 0.05 spins/Rc1
Fe
molecule) was detectable. Recent Mo
¨ssbauer studies con-
firmed that both the FeFe cofactor and the P-cluster are
diamagnetic in the dithionite-reduced state and that the
cofactor contains four Fe
II
- and four Fe
III
-centers [21]. For
the analogous, dithionite-reduced state of the FeMo-
cofactor, the presence of four Fe
II
but only three Fe
III
centers in addition to the Mo
IV
center has been postulated
[35]. Thus, the FeFe-cofactor may be (formally) regarded as
a FeMo-cofactor molecule in which molybdenum has been
replaced by an Fe
III
center [21].
When the FeFe protein was oxidized with K
3
[Fe(CN)
6
],
in a stepwise fashion similar to that described for the MoFe-
protein, several novel EPR signals were detected. The two
most prominent signals (both S¼1/2) have already been
partially characterized [8]. One of these is a very narrow
rhombic signal at g¼2.00, 1.98 and 1.96 (in the following
designated as g¼1.98 signal) and the other, a characteristic
broad signal with an absorption-shaped peak at g¼2.27
and a derivative-shaped feature at g¼2.06 (in the following
termed g¼2.27 signal). The two signals are depicted in
spectra 3 and 4 of Fig. 1 and directly compared to the most
characteristic S¼1/2 signals of the reference system (the
oxidized MoFe protein), that have been attributed to P
1+
Fig. 3. pH-dependent occurrence of the axial EPR signal (g
||
¼2.00,
g
^
¼1.90) resulting from the partially oxidized MoFe protein. Two
samples of the redox titration, both of the potential region where the
rhombic P
1+
signal shows maximal intensity ()120 to )90 mV), were
thawed and adjusted to pH 6.4 and 8.4, respectively, with a concen-
trated three-component buffer system (0.87
M
Bistris, 0.44
M
Hepps,
0.44
M
Ches) according to [37]. The spectrum of the pH 7.4 sample
represents the original spectrum. All spectra were recorded at 16 K and
100 mW. Inset: difference spectrum (spectrum pH 8.4 )spectrum
pH 6.4) depicting the axial signal.
1654 S. Siemann et al. (Eur. J. Biochem. 269)ÓFEBS 2002


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)