
BioMed Central
Page 1 of 5
(page number not for citation purposes)
World Journal of Surgical Oncology
Open Access
Case report
Primary carcinoid tumors of the liver
Gary Schwartz*1, Agnes Colanta2, Harold Gaetz2, John Olichney3 and
Fadi Attiyeh1
Address: 1Department of Surgery, 1000 10th Avenue, Suite 2B, New York, NY, 10019, USA, 2Department of Pathology, St. Luke's-Roosevelt
Hospital Center, 1000 10th Avenue, 1st Floor, New York, NY, 10019, USA and 3Department of Hematology-Oncology, St. Luke's-Roosevelt
Hospital Center, 350 West 58th Street, New York, NY, 10019, USA
Email: Gary Schwartz* - gsschwartz@gmail.com; Agnes Colanta - acolanta@chpnet.org; Harold Gaetz - hgaetz@chpnet.org;
John Olichney - jolichney@chpnet.org; Fadi Attiyeh - fattiyeh@chpnet.org
* Corresponding author
Abstract
Background: Primary carcinoid tumors of the liver are uncommon and rarely symptomatic. The
diagnosis of primary hepatic etiology requires rigorous workup and continued surveillance to
exclude a missed primary.
Case Presentation: We present a case of a 62-year-old female with a primary hepatic carcinoid
tumor successfully resected, now with three years of disease-free follow-up. We present a review
of the current literature regarding the diagnosis, pathology, management, and natural history of this
disease entity.
Conclusion: Primary carcinoid tumors of the liver are rare, therefore classifying their nature as
primary hepatic in nature requires extensive workup and prolonged follow-up. All neuroendocrine
tumors have an inherent malignant potential that must be recognized. Management remains surgical
resection, with several alternative options available for non-resectable tumors and severe
symptoms. The risk of recurrence of primary hepatic carcinoid tumors after resection remains
unknown.
Background
Although carcinoid tumors can be found throughout the
body, 90% occur within the gastrointestinal tract [1]. They
preferentially metastasize to the liver and occasionally (<
10%) cause the carcinoid syndrome by secretion of serot-
onin and its precursors, as well as other vasoactive sub-
stances [2]. Primary carcinoid tumors of the liver are
exceedingly rare, with only about 60 cases reported in the
current literature. Meticulous follow-up is necessary to
rule out an occult extrahepatic malignancy with hepatic
metastasis to confirm the primary nature of hepatic carci-
noids.
Case presentation
EG is a 62-year-old female who presented with right upper
quadrant abdominal pain, intermittent in timing and dull
in nature, not related to oral intake and not associated
with nausea or vomiting. Her past medical history
included hypertension, irritable bowel syndrome, oste-
oarthritis, and a history of recurrent bilateral lower
extremity deep venous thrombosis on Warfarin. On phys-
ical exam there were no abdominal scars, normal bowel
sounds on auscultation, minimal right upper quadrant
tenderness to palpation, no rebound tenderness or guard-
ing, no hepatomegaly and a negative Murphy's sign. Her
Published: 27 August 2008
World Journal of Surgical Oncology 2008, 6:91 doi:10.1186/1477-7819-6-91
Received: 21 June 2008
Accepted: 27 August 2008
This article is available from: http://www.wjso.com/content/6/1/91
© 2008 Schwartz et al; licensee BioMed Central Ltd.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Surgical Oncology 2008, 6:91 http://www.wjso.com/content/6/1/91
Page 2 of 5
(page number not for citation purposes)
laboratory studies were significant for a GGT of 162 U/L
(normal 5–80 U/L), with otherwise normal liver function
tests. Tumor markers were negative, with an AFP of 3.1 ng/
ml.
Diagnostic imaging included an abdominal ultrasound
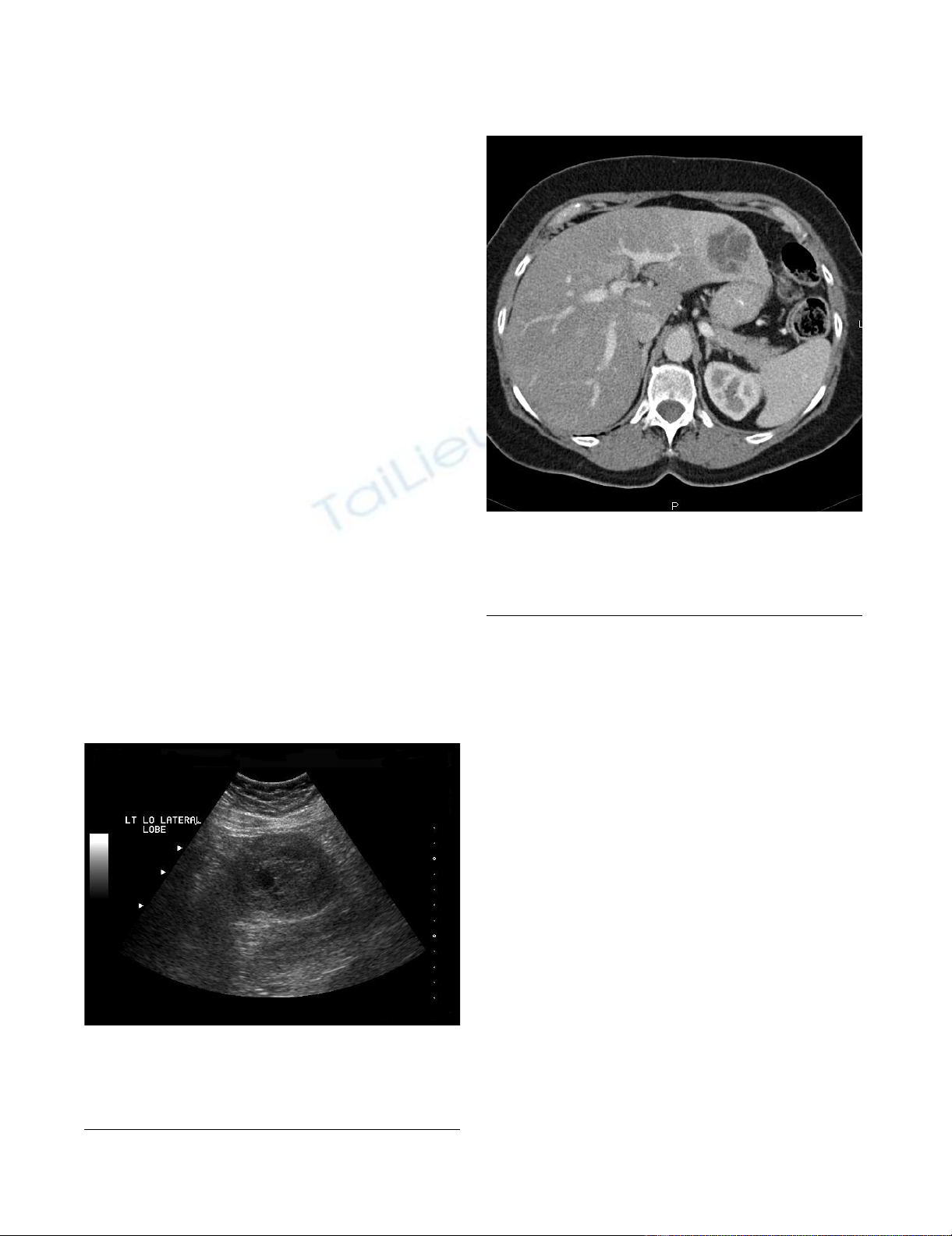
(Figure 1) which revealed a heterogeneous solid mass in
the lateral segment of the left hepatic lobe measuring 6.3
× 5.3 × 5.0 cm. A CT scan with intravenous contrast was
obtained which revealed a 4.9 × 4.9 cm enhancing, poorly
marginated mass in segment II of the liver, with no other
intra-abdominal masses or lymphadenopathy (Figure 2).
A CT-guided biopsy was performed which yielded scant
tissue with poorly cohesive cells arranged in papillae. PAS-
D stain showed focal, small mucin droplets in some cells.
Immunohistochemistry was positive for CEA and CK-7
and negative for calretinin, CDX-2, CK-20, Muc-2 and
Muc-6. The limited sample was diagnosed as papillary
adenocarcinoma, favoring metastasis, on the basis of mor-
phology, special stain results and immunoprofile. How-
ever, a second panel of immunohistochemical stains for
synaptophysin, CD56 and chromogranin were performed
on the biopsy specimen. The tumor cells were negative for
chromogranin but expressed synaptophysin and CD56,
consistent with the immunoprofile of a neuroendocrine
tumor (NET).
Further workup for a primary tumor or other metastatic
sites included a negative CT scan of the chest, upper and
lower gastrointestinal endoscopy, and a Technetium-99m
bone scan. The decision was made to resect the hepatic
tumor.
An uncomplicated left lateral segmentectomy (II & III)
and cholecystectomy were performed. No peritoneal car-
cinomatosis was noted upon exploration. The postopera-
tive course was uneventful and she was discharged home
on the fourth postoperative day.
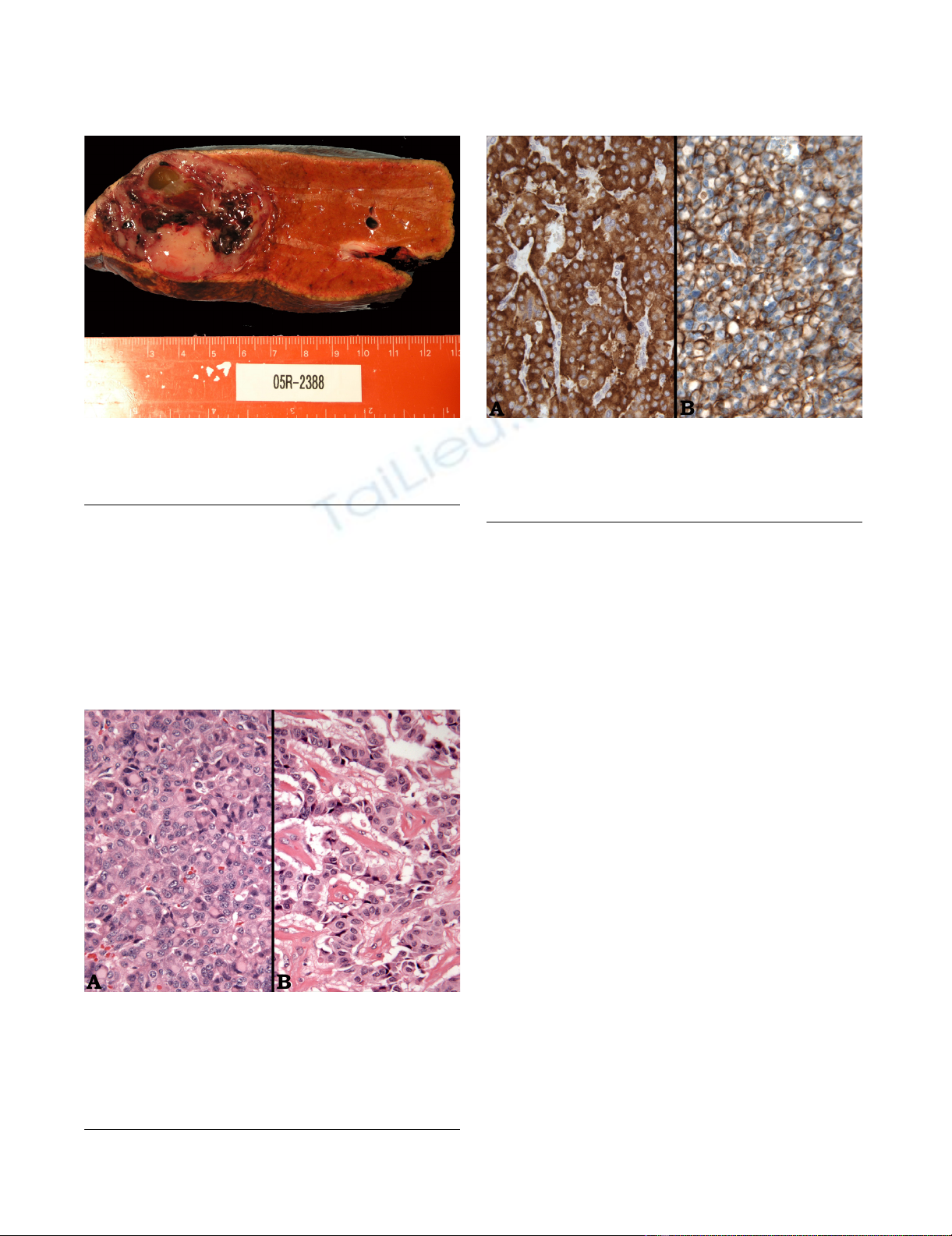
Grossly, the tumor measured 5.2 × 5.0 × 5.0 cm and had
a tan gray, soft, fish-fleshy cut surface (Figure 3). Although
there was a focal infiltrative edge, it was well-circum-
scribed and located 5.9 cm away from the resection mar-
gin. The tumor consisted of approximately 40% solid
areas and 60% hemorrhagic and cystic degenerative areas.
There were no satellite nodules. Surgical margins were
negative for malignancy, including the left hepatic artery,
vein, duct, gallbladder and portal lymph nodes.
Microscopically, the tumor consisted predominantly of
solid sheets and organoid nests of uniform, intermediate-
sized, polyhedral cells (Figure 4A) in a vascular stroma.
Other areas showed a trabecular arrangement of these
cells with focal stromal hyalinization (Figure 4B); cystic
areas were also present. Cytologically, the tumor cells had
a moderate amount of eosinophilic cytoplasm with peri-
nuclear eosinophilic inclusions and round to oval nuclei
Ultrasound of the abdomen; Ultrasound of the abdomen depicting a 6.3 × 5.3 × 5.0 heterogenous solid mass in the lat-eral segment of the left lobe of the liverFigure 1
Ultrasound of the abdomen; Ultrasound of the abdo-
men depicting a 6.3 × 5.3 × 5.0 heterogenous solid
mass in the lateral segment of the left lobe of the
liver.
CT scan of the abdomen and pelvis; CT scan of the abdomen and pelvis with IV contrast demonstrates a 4.9 × 4.9 cm enhancing, poorly marginated mass in segment II of the liverFigure 2
CT scan of the abdomen and pelvis; CT scan of the
abdomen and pelvis with IV contrast demonstrates a
4.9 × 4.9 cm enhancing, poorly marginated mass in
segment II of the liver.
World Journal of Surgical Oncology 2008, 6:91 http://www.wjso.com/content/6/1/91
Page 3 of 5
(page number not for citation purposes)
with vesicular to finely granular chromatin. There were no
areas of necrosis, and mitoses were infrequent.
Immunohistochemistry was consistent with the immuno-
profile of the biopsy specimen, i.e. positive staining for
synaptophysin (Figure 5A) and CD56 (Figure 5B) and
negative staining for chromogranin. Additionally, there
was immunoreactivity for epithelial markers CK-7, CAM
5.2 and pancytokeratin AE1/AE3. There was negative
staining for HEPT, CA19.9 and TTF-1, thus ruling out
hepatocellular carcinoma, metastatic carcinoma from the
gastrointestinal tract and metastatic lung carcinoma,
respectively. The histomorphologic features coupled with
the immunohistochemical results supported the diagno-
sis of a carcinoid tumor/low grade NET.
Follow-up over the subsequent three years included CT
scans of the abdomen at six month intervals. To date, no
recurrent or metastatic disease has been identified. She
remains symptom free and in good health.
Discussion
A total of sixty cases of primary hepatic carcinoid have
been reported, with the largest series being eight patients
[3], with long-term follow-up ranging from two to eleven
years. Of the reported cases, there is a wide range of age at
presentation and there does not seem to be gender pre-
dominance. There is no apparent association with cirrho-
sis or preexisting liver disease.
Primary hepatic carcinoid tumors may be an incidental
finding or can present with severe symptoms including
abdominal pain, jaundice, palpable right upper quadrant
mass, carcinoid syndrome [4], carcinoid heart disease [5],
and Cushing's Syndrome [6]. Less than 10% of gastroin-
testinal carcinoids present with the carcinoid syndrome
and when the syndrome is present it is almost always
associated with hepatic metastasis. Interestingly, the syn-
drome is rarely present in primary hepatic carcinoid
tumors, with only two reported cases [4,5].
Gross image of the specimen; The specimen was measured at 5.2 × 5.0 × 5.0 cm and had a tan gray, soft, fish-fleshy cut sur-faceFigure 3
Gross image of the specimen; The specimen was
measured at 5.2 × 5.0 × 5.0 cm and had a tan gray,
soft, fish-fleshy cut surface.
Microscopic image of the specimen; The tumor consisted of solid sheets and organoid nests of uniform, intermediate-sized, polyhedral cells in a vascular stroma (Image A) as well as areas of trabecular arrangement with focal stromal hyalini-zation (Image B)Figure 4
Microscopic image of the specimen; The tumor con-
sisted of solid sheets and organoid nests of uniform,
intermediate-sized, polyhedral cells in a vascular
stroma (Image A) as well as areas of trabecular
arrangement with focal stromal hyalinization (Image
B).
Immunohistochemistry of the resected specimen; Immuno-histochemistry was positive for synaptophysin (Image A) and CD56 (Image B), consistent with a NETFigure 5
Immunohistochemistry of the resected specimen;
Immunohistochemistry was positive for synapto-
physin (Image A) and CD56 (Image B), consistent
with a NET.

World Journal of Surgical Oncology 2008, 6:91 http://www.wjso.com/content/6/1/91
Page 4 of 5
(page number not for citation purposes)
Imaging studies of any hepatic mass should begin with
ultrasound and a triple-phase CT scan. One report sup-
ports the use of contrast-enhanced ultrasound, although
there is limited experience with that modality [7]. MRI is
increasingly being used, with improved visualization of
carcinoid tumors on T2-weighted images [8]. Additional
information can be gained from nuclear medicine imag-
ing scans, specifically utilizing Technetium-99m isotopes,
as was done with our patient [9]. Finally, if carcinoid is
diagnosed postoperatively on histopathology, workup for
a primary gastrointestinal site should continue with upper
and lower gastrointestinal endoscopy, if these were not
performed preoperatively.
The differentiation between primary and secondary NETs
of the liver is not possible by histology alone, although a
centrally located solitary tumor may suggest a primary [3].
Additionally, some epithelial tumors (e.g. well-differenti-
ated hepatocellular carcinoma, adenocarcinomas and
other neoplasms) may exhibit a NET-like morphology. In
such cases, immunohistochemical staining for neuroen-
docrine markers (e.g. chromogranin, synaptophysin,
CD56) should be performed to establish the cell of origin.
However, it should be noted that most laboratories,
including our own, use chromogranin A monoclonal anti-
body to stain for NETs, therefore NETs expressing chrom-
ogranin B may be non-reactive with this antibody, as was
the case with our specimen.
All neuroendocrine tumors have malignant potential. As
such, some authors recommend using the terms "low-
grade neuroendocrine tumor," "well-differentiated neu-
roendocrine tumor," "well-differentiated endocrine
tumor" or "grade I neuroendocrine carcinoma" instead of
"carcinoid tumor" to emphasize their biologic behavior.
The value of the term "neuroendocrine tumor" reflects a
particular phenotype that may respond to specific targeted
therapies [10].
Despite the classic low-grade cytoarchitectural morphol-
ogy present in this patient's tumor, its large size (5.2 cm in
greatest dimension) and focally infiltrative border are
worrisome. As a general principle, NETs smaller than 1.0
cm, in any anatomic location, usually behave in an indo-
lent fashion with only rare recurrences or distant spread
while those larger than 2.0 cm are usually more aggressive
[11]. However, this parameter may not be as important in
primary hepatic NETs when it is noted that some of the
reported cases have tumors ranging in size from 3.0–16
cm and six of eight patients have remained disease-free
after follow-up of more than three years [3]. On the other
hand, there are reported cases with sizes ranging from
8.2–26 cm where three of five patients died as early as
seven months post-operatively [12]. The large size of the
tumors in this particular series was surmised to be the
cause of the unfavorable outcome.
After the appropriate workup of a hepatic mass, initial
management is surgical resection when possible. Extent of
resection is determined by location and size of the
tumor(s), with multicentric bilobar disease often preclud-
ing resection. When this is the case, alternative therapies
include radiofrequency ablation [7], hepatectomy with
transplantation [3], selective hepatic artery embolization
[13], regional or systemic chemotherapy, and intravenous
octreotide infusion for symptomatic relief. The limited
experience with this disease entity makes current recom-
mendations of management difficult. Traditional
approaches to hepatic tumors are employed at the discre-
tion of the treating surgeons, gastroenterologists, inter-
ventional radiologists, and oncologists.
The rigorous follow-up and frequent monitoring of
patients with hepatic carcinoid also serves as screening for
recurrent disease. Recurrences have been reported as early
as one year postoperatively and as late as thirteen years,
and can occur in the liver or in regional lymph nodes [14-
16]. Distant metastasis without primary hepatic recur-
rence has not been reported.
Conclusion
Carcinoid tumors involving the liver are common, but
primary hepatic carcinoid tumors are rare. Classification
as a primary hepatic tumor requires extensive workup and
prolonged follow-up. Regardless of their size, location,
and degree of differentiation, NETs have an inherent
malignant potential that must be recognized. Manage-
ment remains surgical resection, with several alternative
options available for non-resectable tumors and severe
symptoms. The risk of recurrence of primary hepatic carci-
noid tumors after resection remains unknown.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
GS drafted the case presentation and literature review sec-
tions of this manuscript. AC and HG reviewed the speci-
mens and drafted the review of the pathological findings
associated with this disease entity. FA and JO were the pri-
mary physicians diagnosing, treating, and currently fol-
lowing the referenced patient.
Acknowledgements
Written informed consent was obtained from the patient for publication of
this case report and the accompanying images. A copy of the written con-
sent is available for review by the Editor-in-Chief of this journal.

Publish with BioMed Central and every
scientist can read your work free of charge
"BioMed Central will be the most significant development for
disseminating the results of biomedical research in our lifetime."
Sir Paul Nurse, Cancer Research UK
Your research papers will be:
available free of charge to the entire biomedical community
peer reviewed and published immediately upon acceptance
cited in PubMed and archived on PubMed Central
yours — you keep the copyright
Submit your manuscript here:
http://www.biomedcentral.com/info/publishing_adv.asp
BioMedcentral
World Journal of Surgical Oncology 2008, 6:91 http://www.wjso.com/content/6/1/91
Page 5 of 5
(page number not for citation purposes)
References
1. Caplin ME, Buscombe JR, Hilson AJ, Jones AL, Watkinson AF, Bur-
roughs AK: Carcinoid tumor. Lancet 1998, 352:799-805.
2. Evers BD: Small Intestine. In Sabiston Textbook of Surgery 17th edi-
tion. Edited by: Townsend CM, Beauchamp RD, Evers BM, Mattox KL.
Philadelphia: Elsevier Saunders Inc.; 2004:1359-1362.
3. Fenwick SW, Wyatt JI, Toogood GJ, Lodge JP: Hepatic resection
and transplantation for primary carcinoid tumors of the
liver. Ann Surg 2004, 239(2):210-219.
4. Mehta DC, Warner RR, Parnes I, Weiss M: An 18-year follow-up
of primary hepatic carcinoid with carcinoid syndrome. J Clin
Gastroenterol 1996, 23(1):60-62.
5. Tohyama T, Matsui K, Kitagawa K: Primary hepatic carcinoid
tumor with carcinoid syndrome and carcinoid heart disease:
a case report of a patient on long-term follow-up. Intern Med
2005, 44(9):958-962.
6. Shah NA, Urusova IA, D'Agnolo A, Colquhoun SD, Rosenbloom BE,
Vener SL, Geller SA, Yiunes M, Lechago J, Heaney AP: Primary
hepatic carcinoid tumor presenting as Cushing's syndrome.
J Endocrinol Invest 2007, 30(4):327-333.
7. Komatsuda T, Ishida H, Furukawa K, Miyauchi T, Heianna : Primary
carcinoid tumor of the liver: report of a case with an empha-
sis on contrast-enhanced ultrasonographic findings. J Clin
Ultrasound 2005, 33(6):302-304.
8. Fujino K, Koito K, Sano S, Takahara T, Nakamura E, Morisaki Y,
Furuya T, Torigoe T, Ishii Y: A primary hepatic carcinoid tumor:
evaluation by computed tomography and magnetic reso-
nance imaging. Radiat Med 1998, 16(5):371-373.
9. Shih WJ, Samayoa L, Shih GL, Milan P: Primary hepatic carcinoid
tumor presenting as a large multicystic lesion of the liver and
on Tc-99m RBC abdominal imaging showing photopenic
areas. Clin Nucl Med 2005, 30(7):530-531.
10. DeLellis RA, Osamura RY: Neuroendocrine tumors: an over-
view. Pathology Case Reviews 2006, 11(6):229-234.
11. Graeme-Cook F: Neuroendocrine tumors of the GI tract and
appendix. In Surgical Pathology of the GI Tract, Liver, Biliary Tract, and
Pancreas 1st edition. Edited by: Odze R. Philadelphia: Elsevier Saun-
ders Inc.; 2003:485-486.
12. Pilichowska M, Kimura N, Ouchi A, Lin H, Mizuno Y, Nagura H: Pri-
mary hepatic carcinoid and neuroendocrine carcinoma: clin-
icopathological and immunohistochemical study of five
cases. Pathol Int 1999, 49(4):318-324.
13. Sano K, Kosuge T, Yamamoto J, Shimada K, Takayama T, Yamsaki S,
Makuuchi M: Primary hepatic carcinoid tumors confirmed
with long-term follow-up after resection. Hepatogastroenterol-
ogy 1999, 46(28):2547-2550.
14. Abdel Wahab M, Fathy O, Elghwalby N, Sultan A, Mostafa M, El-Baz
M, Elsaadany M, Elshobary M, Ezzat F: Primary hepatic carcinoid
tumor: one Egyptian center experience. Hepatogastroenterology
2006, 53(67):33-38.
15. Iimuro Y, Deguchi Y, Ueda Y, Tanaka A, Iwasa Y, Ishihara M, Mizuta
K, Yamamoto Y, Ikai I, Shimahara Y, Yamaoka Y: Primary hepatic
carcinoid tumor with metachronous lymph node metastasis
after long-term follow up. J Gastroenterol Hepatol 2002,
17(10):1119-1124.
16. Nishimori H, Tsuji K, Miyamoto N, Sakurai Y, Mitsui S, Kang JH, Yosh-
ida M, Nomura M, Fuminori I, Ishiwatari H: Recurrence of primary
hepatic carcinoid tumor in the remnant liver 13 yr after
resection. Int J Gastrointest Cancer 2005, 35(2):147-151.


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)