
MINIREVIEW
Mechanisms of obesity and related pathologies: Role of
apolipoprotein E in the development of obesity
Kyriakos E. Kypreos
1
, Iordanes Karagiannides
2
, Elisavet H. Fotiadou
1
, Eleni A. Karavia
1
,
Maria S. Brinkmeier
1
, Smaragda M. Giakoumi
1
and Eirini M. Tsompanidi
1
1 Department of Medicine, Pharmacology Unit, University of Patras Medical School, Rio, Greece
2 Department of Medicine, David Geffen School of Medicine, UCLA, Los Angeles, CA, USA
Introduction
Apolipoprotein E (ApoE) is a major protein of the
lipid and lipoprotein transport system mainly
involved in the metabolism of dietary lipids and the
removal of atherogenic lipoproteins, such as chylomi-
cron remnants and very low density lipoproteins
(VLDL), from the circulation [1,2]. In humans,
ApoE is a polymorphic 34.5 kDa glycoprotein
synthesized primarily by the liver, although it is also
synthesized by other tissues, such as brain and
adipose tissue. Human ApoE has three natural
isoforms, ApoE2, ApoE3 and ApoE4 [3]. These
isoforms differ in their amino acid compositions at
positions 112 and 158, where ApoE2 has Cys at
both sites, ApoE4 has Arg at both sites, and ApoE3
has Cys112 and Arg158 [3]. Epidemiological studies
have linked ApoE4 to elevated LDL cholesterol
levels and an increased risk of the development of
cardiovascular disease [4,5].
Lipoprotein-bound ApoE is the natural ligand for
the LDL-receptor (LDLr) [6,7], which is the main
receptor involved in the clearance of ApoE-containing
lipoproteins in vivo [8]. After a lipid-rich meal, dietary
Keywords
ApoE receptors; ApoE3
knock-in
mice;
ApoE4
knock-in
mice; ApoE-deficient mice;
apolipoprotein E; glucose intolerance; insulin
resistance; metabolic syndrome; obesity
Correspondence
K. E. Kypreos, Department of Medicine,
University of Patras Medical School,
Pharmacology Unit, Panepistimioupolis, Rio,
TK 26500, Greece
Fax: +30 2610994720
Tel: +30 2610969120
E-mail: kkypreos@med.upatras.gr
(Received 18 February 2009, revised 1
August 2009, accepted 11 August 2009)
doi:10.1111/j.1742-4658.2009.07301.x
Apolipoprotein E is a polymorphic glycoprotein in humans with a molecu-
lar mass of 34.5 kDa. It is a component of chylomicron remnants, very
low density lipoprotein, low density lipoprotein and high density lipopro-
tein, and is primarily responsible for maintaining plasma lipid homeostasis.
In addition to these well-documented functions, recent studies in experi-
mental mouse models, as well as population studies, show that apolipo-
protein E also plays an important role in the development of obesity and
insulin resistance. It is widely accepted that disruption in homeostasis
between food intake and energy expenditure, and the subsequent deposition
of excess fatty acids into fat cells in the form of triglycerides, leads to the
development of obesity. Despite the pivotal role of obesity and dyslipide-
mia in the development of the metabolic syndrome and heart disease, the
functional interactions between adipose tissue and components of the lipo-
protein transport system have not yet been investigated thoroughly. In this
minireview, we focus on the current literature pertinent to the involvement
of apolipoprotein E in the development of pathologies associated with the
metabolic syndrome.
Abbreviations
ABCA1, ATP-binding cassette A1; ApoE, apolipoprotein E; ApoE
)⁄)
, ApoE-deficient; HDL, high density lipoprotein; LCAT, lecithin:cholesterol
acyl transferase; LDLr, low density lipoprotein receptor; LDLr
)⁄)
, LDLr-deficient; LpL, lipoprotein lipase; LRP1, LDLr related protein 1; VLDL,
very low density lipoprotein; VLDLr, very low density lipoprotein receptor.
5720 FEBS Journal 276 (2009) 5720–5728 ª2009 The Authors Journal compilation ª2009 FEBS
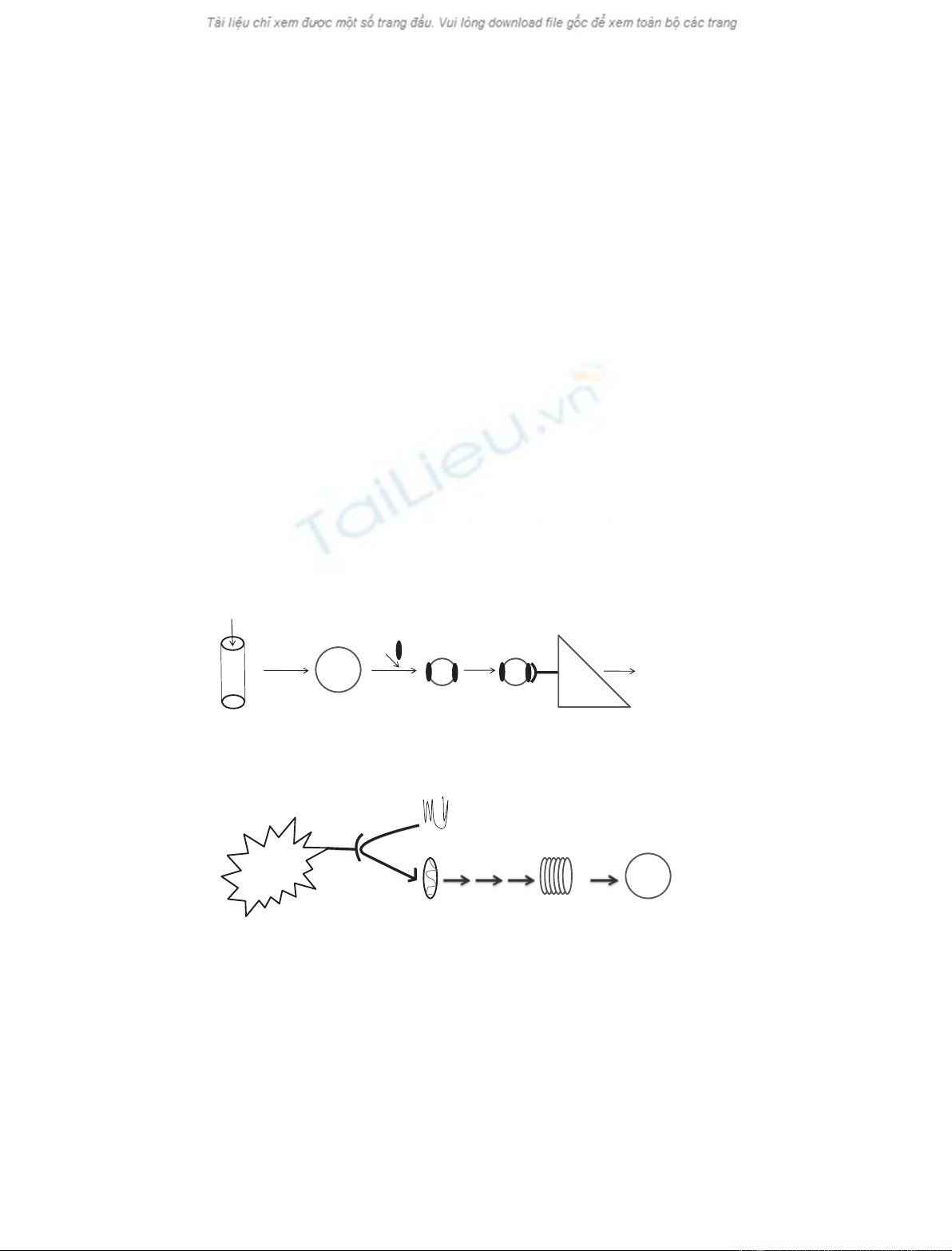
lipids are packaged into chylomicrons, which, subse-
quent to partial lipolysis by lipoprotein lipase (LpL),
are converted into chylomicron remnants and acquire
ApoE [2] (Fig. 1A). Then, lipid bound ApoE interacts
with the LDLr, which mediates the removal of ApoE-
containing atherogenic lipoproteins from the circula-
tion (Fig. 1A). Mutations in ApoE or LDLr that
prevent their physical interactions are associated with
high plasma cholesterol levels and predispose to pre-
mature atherosclerosis in humans and experimental
animals [9,10].
In addition, ApoE also promotes cholesterol efflux
[11] and the de novo biogenesis of spherical ApoE-con-
taining high density lipoprotein (HDL)-like particles
with the participation of the lipid transporter ATP bind-
ing cassette A1 (ABCA1) and the plasma enzyme leci-
thin:cholesterol acyl transferase (LCAT) (Fig. 1B) [12].
Thus, ApoE may also contribute to the maintenance of
plasma and tissue cholesterol homeostasis and the pro-
tection from atherosclerosis [13–20] via mechanisms that
are independent of its interactions with the LDLr [18].
It is widely accepted that disruption in the homeo-
stasis between food intake and energy expenditure,
and the subsequent deposition of excess fatty acids
into fat cells in the form of triglycerides, leads to the
development of obesity [21]. A lipid-rich diet and sed-
entary lifestyle, physical inactivity and an imbalance
in caloric load are the most common contributors to
the development of central obesity and the metabolic
syndrome [22,23]. Aging, hormonal imbalance and
genetic predisposition may also contribute to obesity
[24–35].
Epidemiological and population studies have
established a direct correlation between obesity and
the development of cardiovascular disease [36,37].
Despite the pivotal role of obesity and dyslipidemia in
the development of the metabolic syndrome and heart
disease, the functional interactions between adipose tis-
sue and the lipid and lipoprotein transport system have
only recently started to be investigated.
ApoE in adipocyte differentiation and
lipid loading
In vitro experiments using cultures of primary prea-
dipocytes, adipocytes, adipose tissue explants or
Peripheral tissues
or liver
ABCA1
NC
Plasma
apoE
Minimally
lipidated
apoE
Discoidal
apoE-HDL
LCAT
Spherical
apoE-HDL
Chylomicrons
ApoE-containing
chylomicron
remnants
LpL-mediated
lipolysis
Interactions of
remnant-bound apoE
with LDLr
Secretion
of lipid-rich
chylomicrons
in the
circulation
Clearance of dietary
lipids from
the circulation
Lipid-rich
meal
Intestine
ApoE LDLr
1
2
4
A
B
3
Fig. 1. (A) Summary of the role of ApoE in the clearance of chylomicron remnants and VLDL from the circulation. Dietary lipids are packaged
into chylomicrons, which are then partially lipolyzed by plasma lipoprotein lipase on the surface of vascular endothelial cells. Subsequent to
lipolysis, chylomicrons acquire ApoE and are converted into chylomicron remnants. ApoE-containing chylomicron remnants are then taken up
by the liver and other peripheral tissues mainly via the LDLr, which appears to be the major physiological receptor for remnant clearance. (B)
Depicting the pathway of de novo biogenesis of ApoE-containing HDL with the participation of the lipid transporter ABCA1 and plasma
enzyme LCAT. Minimally lipidated ApoE in plasma interacts with ABCA1 (step 1) that is present in the liver or other peripheral tissues. This
interaction promotes the lipidation of ApoE (step 2), which is then converted into a discoidal HDL-like particle through a sequence of steps
that are not yet well understood (step 3). Then, ApoE containing discoidal HDL-like particles are converted into spherical HDL by the action
of the plasma enzyme LCAT (step 4).
K. E. Kypreos et al. ApoE and obesity
FEBS Journal 276 (2009) 5720–5728 ª2009 The Authors Journal compilation ª2009 FEBS 5721

3T3-L1 cells provide some information on the role of
ApoE in preadipocyte differentiation and on ApoE
expression from mature adipocytes.
A study by Chiba et al. [38] provided the first direct
evidence that lipid-bound ApoE promotes preadipo-
cyte differentiation in a dose-dependent manner. Using
bone marrow stromal cells from ApoE-deficient
(ApoE
)⁄)
) mice and 3T3-L1 cells, these investigators
showed that ApoE-deficient VLDL failed to induce
adipogenesis, whereas normal VLDL promoted differ-
entiation of these cells into fat cells. Incubation of
ApoE-deficient VLDL with recombinant human ApoE
partially restored its ability to stimulate adipogenesis,
whereas the selective removal of ApoE from VLDL by
trypsin abolished the adipogenic activity of human
VLDL. When tetrahydrolipstatin, a potent lipoprotein
lipase inhibitor, was used in these experiments, it did
not alter the ability of ApoE-containing VLDL to pro-
mote adipogenesis, suggesting that hydrolysis of
VLDL triglycerides does not play a major role in the
adipogenic effects of ApoE-containing VLDL. Simi-
larly, individual lipid components of the VLDL or free
fatty acids alone induced the expression of adipocyte-
specific genes but failed to generate adipocytes filled
with large lipid droplets, and this finding was inter-
preted as partial adipogenesis compared to the effects
of ApoE-containing VLDL.
Along the same lines, a study by Huang et al. [39]
suggested that the endogenous expression of ApoE
promotes lipid accumulation and adipocyte differentia-
tion in cell cultures. Specifically, adipocytes isolated
from ApoE-deficient mice contained lower levels of tri-
glyceride and free fatty acids compared to adipocytes
isolated from wild-type mice, and these differences
were also maintained in cultured adipocytes derived
from preadipocytes. During incubation with ApoE-
containing triglyceride-rich lipoproteins, ApoE-defi-
cient adipose tissue accumulated less triglycerides than
adipose tissue isolated from wild-type mice. Similarly,
a lack of ApoE expression in primary cultured adipo-
cytes led to changes in the expression of genes involved
in the metabolism ⁄turnover of fatty acids and the tri-
glyceride droplet, whereas peroxisome proliferator-acti-
vated receptor gamma-mediated changes in lipid
content and gene expression were markedly altered in
cultured ApoE-deficient adipocytes. Interestingly, when
human ApoE3 was expressed by adenovirus-mediated
gene transfer in cultured adipocytes from ApoE-defi-
cient mice, it promoted the accumulation of triglyce-
rides and fatty acids in the infected cells. This finding
is in agreement with a study by Zechner et al. [40] who
showed that ApoE expression in differentiating 3T3-L1
cells increases linearly with time in differentiation,
whereas the inhibition of lipid accumulation in differ-
entiated cells by biotin deprivation decreased ApoE
expression.
Interestingly, another set of experiments conducted
by Huang et al. [41] suggested that ApoE expression in
adipocytes was affected by the feeding state of the
mice that the tissue was derived from. ApoE expres-
sion was induced by fasting, whereas diet-induced
obesity or hyperphagia was associated with the
reduced expression of ApoE in the adipose tissue.
Because other studies showed that ApoE-expression in
the adipose tissue promoted lipid accumulation and
adipocyte differentiation [39], one interpretation of the
results obtained by Huang et al. [41] is that intrinsic
defense mechanisms in adipose tissue limit adipogene-
sis by reducing the expression of ApoE in the fed state.
Certainly, additional studies are required to determine
the role of adipocyte-synthesized ApoE, and to distin-
guish between the functions of peripherally expressed
ApoE versus adipocyte expressed ApoE.
Studies in experimental mouse models
Despite the differences in anatomy, pathology, physiol-
ogy and metabolism between mice and humans, studies
in mice during the last few decades have provided
important leads with respect to the pathogenesis and
genetics of human metabolic diseases, including obes-
ity. A number of studies in experimental mouse models
have provided a definitive link between ApoE and
obesity.
Work by Chiba et al. [38] demonstrated that leptin
deficient (ob ⁄ob) mice that are also deficient in apoE
(ob ⁄ob ·ApoE
)⁄)
) did not show an increased body
weight or an increased amount of adipose tissue when
fed a high-fat ⁄high-cholesterol diet, despite an increase
in their plasma VLDL levels. By contrast, control
ob ⁄ob mice fed a high-fat ⁄high-cholesterol diet for the
same period of time showed an increased body weight
and amount of adipose tissue, suggesting that ApoE is
a key modulator of adipogenesis in vivo.
In agreement with that study, Huang et al. [39]
reported that ApoE
)⁄)
mice have less body fat content
and smaller adipocytes compared to wild-type
C57BL ⁄6 controls. A study by Hofmann et al. [42] fur-
ther extended this observation by showing that
ApoE
)⁄)
mice fed a high-fat-high-sucrose diabetogenic
diet for 24 weeks were resistant to diet-induced obesity
and exhibited improved glucose tolerance and uptake
by muscle and brown adipose tissue, whereas their
plasma insulin levels were lower compared to control
wild-type C57BL ⁄6 mice. The reduced body weight
and improved glycemic control of the ApoE
)⁄)
mice
ApoE and obesity K. E. Kypreos et al.
5722 FEBS Journal 276 (2009) 5720–5728 ª2009 The Authors Journal compilation ª2009 FEBS

was accompanied by impaired plasma triglyceride
clearance and lipid uptake by adipose tissue. Direct
calorimetry studies did not reveal any significant differ-
ences in energy expenditure and respiratory quotient
between ApoE
)⁄)
and wild-type C57BL ⁄6 mice fed a
high-fat, high-sucrose diet for 24 weeks, suggesting
that, in the absence of ApoE, decreased plasma lipid
delivery to insulin sensitive tissues improves insulin
sensitivity and prevents the development of diet
induced obesity.
Using an approach similar to Chiba et al. [38], Gao
et al. [43] established that ApoE deficiency in Ay
⁄+
mice prevented the development of obesity, with
decreased fat accumulation in the liver and adipose tis-
sues. Ay (also known as lethal yellow) is a mutation at
the mouse agouti locus in chromosome 2 that results
in a number of dominant pleiotropic effects, including
a yellow coat color, obesity, an insulin-resistant type II
diabetic condition, and an increased propensity to
develop a variety of spontaneous and induced tumors
[44]. The Ay mutation is the result of a 170 bp deletion
that removes all but the promoter and noncoding first
exon of the Raly gene, which lies in the same transcrip-
tional orientation as agouti and maps 280 kb proximal
to the 3¢end of the agouti gene [44]. Gao et al. [43]
generated ApoE-deficient Ay (ApoE
)⁄)
·Ay
⁄+
) mice
and found that ApoE
)⁄)
·Ay
⁄+
mice exhibited better
glucose tolerance than ApoE
+⁄+
·Ay
⁄+
mice,
whereas insulin tolerance testing and hyperinsulinemic-
euglycemic clamp analysis revealed a marked improve-
ment of insulin sensitivity in ApoE
)⁄)
·Ay
⁄+
mice
compared to ApoE
+⁄+
·Ay
⁄+
mice, despite an
increase in their plasma free fatty acid levels. When
these investigators used adenovirus-mediated gene
expression of ApoE in ApoE
)⁄)
·Ay
⁄+
mice, ApoE
protein expression in the plasma of these mice wors-
ened the glucose tolerance and insulin sensitivity of the
ApoE
)⁄)
·Ay
⁄+
mice, and triggered obesity, indicat-
ing that circulating ApoE is involved in increased
adiposity and obesity-related metabolic disorders. Of
note, the uptake of ApoE-lacking VLDL into the liver
and adipocytes was markedly inhibited, although
adipocytes in ApoE
)⁄)
·Ay
⁄+
mice exhibited normal
differentiation.
In a recent study from our laboratory [45], we
established that ApoE3
knock-in
mice fed the standard
Western-type diet for 24 weeks were more sensitive
to diet-induced obesity and related metabolic dys-
functions than wild-type C57BL ⁄6 mice, whereas
ApoE
)⁄)
mice were resistant to the development of
these conditions. Furthermore, deficiency in the
LDLr resulted in reduced sensitivity towards obesity
in response to a Western-type diet (Harlan-Teklad,
catalogue number TD 88137, Indianapolis, IN,
USA), raising the possibility that the effects of ApoE
may be mediated, at least in part, via its interactions
with the LDLr. Of note, ApoE3
knock-in
mice had
lower steady-state plasma ApoE levels than C57BL ⁄6
mice, establishing that the difference in the ability of
human ApoE3 and murine ApoE to promote obesity
in response to a high-fat diet may be the result of
intrinsic differences between these two peptides. Inter-
estingly, in our experiments, we did not observe sig-
nificant differences in plasma free fatty-acid levels
among mouse groups (ApoE3
knock-in
versus C57BL ⁄6
versus LDLr
)⁄)
versus ApoE
)⁄)
), although previous
studies suggested that increased plasma levels of free
fatty acids are closely associated with obesity-induced
insulin resistance [46,47]. Daily food consumption of
the ApoE3
knock-in
, C57BL ⁄6 and ApoE
)⁄)
mice was
similar among groups, suggesting that different
responses to a Western type diet could not be attrib-
uted to differences in appetite. It is quite interesting
that, in all our experiments, plasma cholesterol levels
correlated inversely with body weight gain and body
fat accumulation. In the ApoE
)⁄)
mice, failure to
clear chylomicron remnants because of a deficiency
in ApoE resulted in a steady increase in plasma cho-
lesterol levels and rendered these mice resistant to
diet-induced obesity. By contrast, in the ApoE3
knock-
in
mice, the efficient catabolism of chylomicron rem-
nants resulted in only slightly elevated plasma choles-
terol levels, but promoted obesity, insulin resistance
and glucose intolerance. Similar to the ApoE3
knock-in
mice, C57BL ⁄6 mice, which express the mouse ApoE,
developed only mild hypercholesterolemia, but
became obese and insulin resistant after consuming a
Western-type diet for 24 weeks. Direct measurements
of dietary lipid delivery to hepatic and adipose tissue
raised the possibility that chylomicron and VLDL
remnants containing the human ApoE3 isoform are
taken up more avidly by adipose tissue than the lipo-
proteins containing mouse ApoE.
There has been much discussion in the medical com-
munity concerning the role of inflammation in obesity.
In particular, although some studies suggest that
inflammation causes obesity, others present data
supporting the idea that inflammation is simply a
metabolic side-effect of the obese state. ApoE is long-
known to be an anti-inflammatory molecule [48], and
a deficiency in ApoE is considered to induce general
inflammation that leads to spontaneous atherosclerosis
in the ApoE
)⁄)
mice [49]. Thus, the resistance of
ApoE
)⁄)
mice to developing diet-induced obesity may
support the theory that inflammation does not trigger
obesity, but rather it is the result of it.
K. E. Kypreos et al. ApoE and obesity
FEBS Journal 276 (2009) 5720–5728 ª2009 The Authors Journal compilation ª2009 FEBS 5723

In our studies, we also found that LDLr
)⁄)
mice
became more obese than ApoE
)⁄)
mice, yet less obese
than C57BL ⁄6 mice, raising the possibility that, in
addition to the LDLr, other ApoE-recognizing recep-
tors may also promote the deposition of postprandial
lipids to adipose tissue, thus contributing to diet-
induced obesity and related metabolic dysfunctions.
Thus, in the absence of LDLr, other ApoE-recognizing
‘scavenger’ receptors, such as LDLr-related protein
(LRP1) and very low density lipoprotein receptor
(VLDLr) may promote, to some extent, delivery of
ApoE-containing chylomicron remnants to adipose tis-
sue. However, in the case of the ApoE
)⁄)
mice that
lack the endogenous ApoE, all these ApoE-mediated
receptor processes may be blocked, and ApoE
)⁄)
mice
become more resistant to body fat gaining compared
to LDLr
)⁄)
mice. Indeed, Hofmann et al. [50] showed
that adipocyte-specific inactivation of the multifunc-
tional receptor LRP1 in mice resulted in delayed post-
prandial lipid clearance, reduced body weight, smaller
fat stores, lipid-depleted brown adipocytes, improved
glucose tolerance and elevated energy expenditure as a
result of enhanced muscle thermogenesis. Furthermore,
inactivation of adipocyte LRP1 resulted in resistance
to dietary fat-induced obesity and glucose intolerance.
In another study by Gourdiaan et al. [51] VLDLr-defi-
cient mice were found to be resistant to diet-induced
obesity when fed a high-fat, high-calorie diet. Thus, it
is possible that, in the absence of LDLr, remnant-
bound ApoE interacts with VLDLr or LRP1 present
on the surface of adipocytes [52,53] to facilitate the
lipolysis of VLDL-triglycerides by LpL [53] and possi-
bly the subsequent uptake of remnant particles by
ApoE-recognizing receptors [50].
In humans, ApoE has three natural isoforms: ApoE2,
ApoE3 and ApoE4 [3]. In vitro receptor binding studies
have established that lipid bound ApoE3 and ApoE4
have a similar affinity for the LDLr, whereas lipid
bound ApoE2 has a much lower affinity [54]. If the
effects of ApoE3 on obesity are mediated solely by its
lipid lowering potential via the LDLr and possibly other
ApoE recognizing receptors, it would be expected that
both ApoE3 and ApoE4 will predispose to a similar
extent to diet-induced obesity and insulin resistance in
mice, whereas ApoE2 may have a much lower potential
to promote these conditions. One study [55] investigated
the contribution of the natural human ApoE3 and
ApoE4 phenotypes in the development of obesity and
other metabolic abnormalities in mice. ApoE3
knock-in
and ApoE4
knock-in
mice were fed Western-type diet for
8 weeks and, during this time, the sensitivity of these
mice towards the development of obesity and glucose
tolerance was assessed. Analysis of total fat content
showed that ApoE3
knock-in
mice had more total and
subcutaneous fat than ApoE4
knock-in
mice at the end of
the 8-week period. However, although ApoE4
knock-in
mice gained 30% less weight during the period on high-
fat diet compared to ApoE3 mice, they showed
impaired insulin-stimulated glucose uptake. Further-
more, epididymal adipocytes derived from ApoE4
knock-
in
mice were larger in size than those derived from
ApoE3
knock-in
mice. When ApoE3 and ApoE4 were
expressed by adenovirus-mediated gene transfer in cul-
tures of ApoE-deficient adipocytes, only ApoE3 expres-
sion was able to significantly induce adiponectin
mRNA expression, and mobilize the glucose transporter
GLUT4, suggesting that ApoE3 but not ApoE4 expres-
sion interferes with insulin sensing pathways. On the
basis of these findings, it was concluded that, even
though ApoE3 expression leads to higher adipose tissue
mass in mice compared to ApoE4, qualitative differ-
ences in the epididymal adipose tissue between the
ApoE3
knock-in
and ApoE4
knock-in
mice contribute to
the accelerated impairment of glucose tolerance in the
ApoE4
knock-in
mice fed a Western-type diet for 8 weeks.
Although this study did not address the question of
how differences in receptor-mediated clearance of
ApoE-containing lipoproteins and possibly holoparticle
uptake may contribute to an ApoE isoform-dependent
sensitivity towards obesity, it raised the interesting pos-
sibility that metabolic dysfunctions such as impaired
glucose tolerance and insulin sensitivity may be the
result of qualitative differences in fat depots present in
mice expressing different ApoE isoforms. Of course,
obesity and its related complications are chronic dys-
functions that develop over long periods of time. It is
possible that 8 weeks on a high-fat diet was too short a
period for ApoE3
knock-in
and ApoE4
knock-in
mice to
develop obesity and its related metabolic dysfunctions.
Thus, in future studies, it would be interesting to inves-
tigate whether the more obesity-prone ApoE3
knock-in
mouse develops as severe or even more severe metabolic
dysfunctions compared to ApoE4
knock-in
mice, when fed
a Western-type diet for 24 weeks or longer.
Shen et al. [56] suggested that brain apoE expression
reduces food intake in rats. Specifically, the intrecere-
broventricular injection of ApoE in rats decreased
their food intake, whereas intrecerebroventricular infu-
sion of ApoE anti-serum stimulated feeding. However,
in previous studies [38,43,45,55] that compared ApoE-
deficient with ApoE-expressing mice, there were no
significant changes in daily food intake between these
mouse groups. One possibility is that the peripheral
effects of ApoE predisposing to obesity in those
studies offset the brain-specific effects that reduced
food-intake in the study by Shen et al. [56].
ApoE and obesity K. E. Kypreos et al.
5724 FEBS Journal 276 (2009) 5720–5728 ª2009 The Authors Journal compilation ª2009 FEBS


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)