
Structural determinants of protein stabilization by solutes
The importance of the hairpin loop in rubredoxins
Tiago M. Pais
1
, Pedro Lamosa
1
, Wagner dos Santos
1
, Jean LeGall
1,2
, David L. Turner
1,3
and Helena Santos
1
1 Instituto de Tecnologia Quı
´mica e Biolo
´gica, Universidade Nova de Lisboa, Portugal
2 Department of Biochemistry, University of Georgia, Athens, GA, USA
3 Department of Chemistry, University of Southampton, UK
In spite of the extensive accumulation of data on pro-
tein structure, the molecular determinants of protein
thermal stability remain elusive. Also, the beneficial
stabilizing effects exerted by various compatible solutes
have been known for a long time, yet the mechanisms
responsible for this stabilization are a matter of intense
discussion [1–4]. One of the reasons for this apparent
lack of success is that many different factors, both
intrinsic and extrinsic, seem to contribute to the ther-
mostability of any given protein [5]. Protein stability
appears as the result of a delicate balance of stabilizing
and destabilizing interactions, with the thermodynamic
Keywords
compatible solutes; hairpin structure; NMR;
rubredoxin; thermostability
Correspondence
H. Santos, Instituto de Tecnologia Quı
´mica
e Biolo
´gica, Universidade Nova de Lisboa,
Apartado 127, 2780-156 Oeiras, Portugal
Fax: +351 21 4428766
Tel: +351 21 4469828
E-mail: santos@itqb.unl.pt
(Received 22 June 2004, revised 7 December
2004, accepted 17 December 2004)
doi:10.1111/j.1742-4658.2004.04534.x
Despite their high sequence homology, rubredoxins from Desulfovibrio
gigas and D. desulfuricans are stabilized to very different extents by com-
patible solutes such as diglycerol phosphate, the major osmolyte in the
hyperthermophilic archaeon Archaeoglobus fulgidus [Lamosa P, Burke A,
Peist R, Huber R, Liu M Y, Silva G, Rodrigues-Pousada C, LeGall J,
Maycock C and Santos H (2000) Appl Environ Microbiol 66, 1974–1979].
The principal structural difference between these two proteins is the
absence of the hairpin loop in the rubredoxin from D. desulfuricans. There-
fore, mutants of D. gigas rubredoxin bearing deletions in the loop region
were constructed to investigate the importance of this structural feature on
protein intrinsic stability, as well as on its capacity to undergo stabilization
by compatible solutes. The three-dimensional structure of the mutant bear-
ing the largest deletion, D17|29, was determined by
1
H-NMR, demonstra-
ting that, despite the drastic deletion, the main structural features were
preserved. The dependence of the NH chemical shifts on temperature and
solute concentration (diglycerol phosphate or mannosylglycerate) provide
evidence of subtle conformational changes induced by the solute. The kin-
etic stability (as assessed from the absorption decay at 494 nm) of six
mutant rubredoxins was determined at 90 C and the stabilizing effect exer-
ted by both solutes was assessed. The extent of protection conferred by
each solute was highly dependent on the specific mutant examined: while
the half-life for iron release in the wild-type D. gigas rubredoxin increased
threefold in the presence of 0.1 mdiglycerol phosphate, mutant D23|29 was
destabilized. This study provides evidence for solute-induced compaction of
the protein structure and occurrence of weak, specific interactions with the
protein surface. The relevance of these findings to our understanding of the
molecular basis for protein stabilization is discussed.
Abbreviations
DGP, diglycerol phosphate; MG, mannosylglycerate; Rd, rubredoxin; RdDd, rubredoxin from Desulfovibrio desulfuricans; RdDg, rubredoxin
from Desulfovibrio gigas.
FEBS Journal 272 (2005) 999–1011 ª2005 FEBS 999

stability of the native state emerging as a small differ-
ence of large numbers [6]. Similarly, the stabilizing
effect conferred by compatible solutes will be the result
of a plethora of direct and ⁄or indirect, weak interac-
tions between the solute (or the changes that the solute
causes in the solvent properties) and the several chem-
ical groups present on the protein surface, rendering
the magnitude of this effect subtly dependent on the
particular solute ⁄protein pair examined and, therefore,
extremely difficult to predict.
One of the strategies used to explore this maze of
interactions and try to rationalize them is to investi-
gate series of homologous proteins in order to unravel
the structural determinants of protein stabilization by
compatible solutes. In a previous study we compared
the action of a compatible solute, diglycerol phosphate
(DGP), on the stability of rubredoxins from three bac-
terial sources [7]. These small metalloproteins display a
wide variation in thermal stability, despite having a
considerable degree of sequence and structural similar-
ity. Typically, rubredoxins are composed of about
52–54 residues and include a three-stranded bsheet, a
metal centre comprising one iron atom tetrahedrally
coordinated by four cysteine sulfur atoms, and a small
hydrophobic core, which is shielded from solvent
access by a hairpin loop [8]. Despite the structural
similarity between rubredoxins, the degree of stabiliza-
tion conferred by DGP was diverse. Although having
almost no effect on the thermal stability of the rubre-
doxin (Rd) from Desulfovibrio desulfuricans (RdDd),
DGP was able to triple the half-life for thermal dena-
turation of the other two rubredoxins examined. RdDd
is the least heat-stable of the several rubredoxins inves-
tigated, and is the only one not stabilized by DGP.
Conversely, the Rd from D. gigas (RdDg) is the most
stable and strongly stabilized by this solute. The main
structural difference between RdDd and other rubre-
doxins is the lack of seven amino acids in the hairpin
loop.
In order to investigate why this structural feature
(the presence of the loop region) seemed to have such
a profound effect on stability and stabilization of ru-
bredoxins, we constructed a series of mutants of RdDg
with different extents of deletion in the original hairpin
loop. The determination of the NMR solution struc-
ture was deemed important, first, to ensure that the
deletion had not substantially altered the protein struc-
ture (except in the loop region); and second, to provide
the structural detail needed to elucidate the molecular
basis of protein stabilization by solutes. Three point
mutants were also studied to assess the importance of
total surface charge or changes in the most exposed
hydrophobic residue.
DGP and mannosylglycerate (MG), two negatively
charged compatible solutes that we isolated from
hyperthermophiles, were used in this study. The effect
of these solutes on the thermal stability of six mutants
was investigated. Moreover, as chemical shifts are
good indicators of changes in protein structure or
dynamics, the changes of the proton chemical shifts
with temperature and solute concentration were ana-
lysed to extract information on protein ⁄solute inter-
actions.
Results
Thermal stability of rubredoxins
Mutant iron rubredoxins show the same characteristic
bands of the UV–visible absorption spectrum as the
native protein with maxima centred at 380, 494 and
570 nm. These bands are bleached, due to the disrup-
tion of the iron centre when the protein undergoes
denaturation. Monitoring the loss of the metal centre
through the decrease in absorbance at 494 nm provides
an expeditious way to evaluate the kinetic stability of
rubredoxins [9–11]. The half-life (t
1⁄2
) for iron release
of the native and mutant rubredoxins was measured at
90 C.
All rubredoxins examined exhibited mono-exponen-
tial behaviour in regard to the decay of absorbance at
494 nm (data not shown). Complete bleaching of spec-
tral features at 380 and 494 nm occurred without for-
mation of detectable precipitates, either from protein
precipitation or insoluble ferric oxides. The spectral
features did not recover on cooling, which indicates
that protein denaturation under these conditions is an
irreversible process, in agreement with previous studies
regarding thermal denaturation of rubredoxins [7,9,11].
Recombinant RdDg presented a half-life for disrup-
tion of the iron centre (t
1⁄2
) of 96 min; all mutations
resulted in a decrease of this parameter. The mutants
bearing deletions in the loop region showed a dramatic
decrease (between 69 and 89%) in their half-lives relat-
ive to the native form (Table 1; Fig. 1). Interestingly,
mutant D23|29 had a half-life comparable with that of
the RdDd, but the two other mutants lost iron at an
even higher rate. The larger the deletion, the shorter
the half-life became, with mutant D17|29 showing the
lowest value for this parameter. In general, single
mutations had a smaller effect on the rate of iron loss,
except for V8N, which showed a rate comparable with
that of mutant D23|29.
The effect induced by DGP in native RdDg was
impressive with at least a threefold increase of the
half-life [7]. However, the effect observed for the
Structural determinants of protein stabilization T. M. Pais et al.
1000 FEBS Journal 272 (2005) 999–1011 ª2005 FEBS
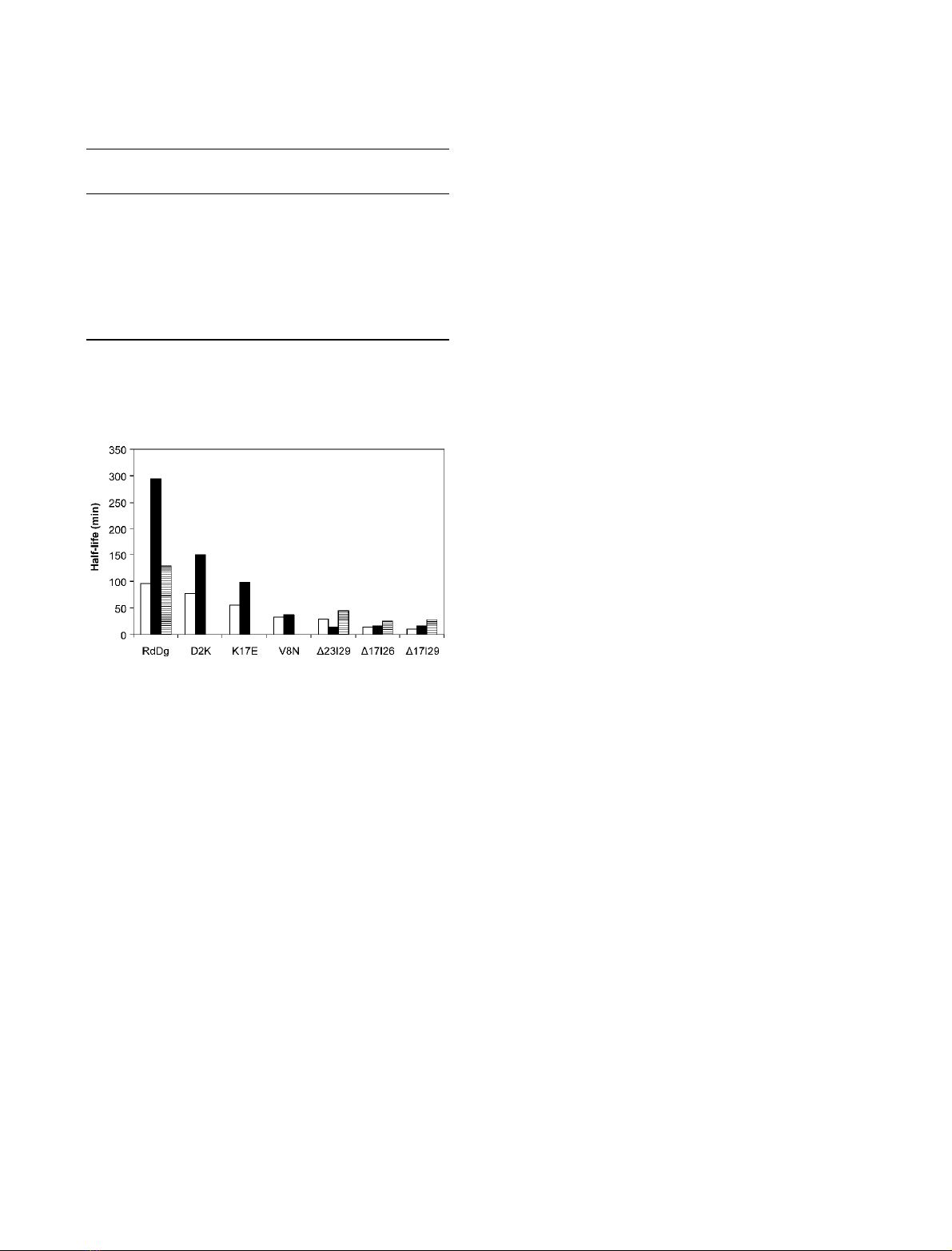
mutant rubredoxins was lower. Mutants D2K, K17E
and D17|29 showed a clear increase in the half-life for
iron loss (between 52 and 94%) but a minor change
was observed with mutants D17|26 and V8N (Table 1;
Fig. 1). Most surprisingly, the half-life of mutant
D23|29 was reduced in the presence of DGP. It is also
interesting to note that, for the point mutants, the
added stabilization follows the intrinsic stability, with
the larger increases occurring in the proteins with
higher intrinsic stability. This trend, however, was not
observed in the case of loop deletions, where the most
stable mutant (with respect to the iron loss), D23|29,
was actually destabilized by addition of DGP. In con-
trast, the presence of MG caused a consistent retarda-
tion on the rates of all rubredoxins examined; in the
case of RdDg the increment of half-life induced by
MG was much lower than that of DGP, but MG
stabilized the deletion-mutants to a much higher
degree, including D23|29. Because K
+
was the counter-
ion for the negative charge of DGP and MG, the
effect of KCl on the rate of iron release was also deter-
mined. We found that KCl had no significant effect on
the half-life of the proteins examined (Table 1).
Structure determination of mutant D17|29 by
NMR
Proton signal assignment was performed using the clas-
sical approach described by Wu
¨thrich [12a]. Analysis
of TOCSY and COSY spectra allowed the identifica-
tion of the spin systems. Sequence-specific assignment
was achieved using NOESY spectra and identifying
connectivities between NH protons and between the
NH and H protons of adjacent spin systems. The spin-
systems for Met1 and Asp19 could not be identified,
probably because mobility of the N-terminus and the
loop region leads to weak signals. Spin diffusion
was taken into account and a value of 6.2% was
used to loosen all NOESY-derived constraints. Stereo-
specific assignments were obtained using preliminary
calculated structures with the aid of program glomsa;
of these, 16 were derived from stereopairs with non-
degenerate chemical shifts and 50 NOESY cross-peaks
could be pseudo-stereospecifically assigned to one or
the other side of the fast-flipping aromatic side chain
rings.
The program indyana was used to generate 500
conformers from which the 20 structures with the low-
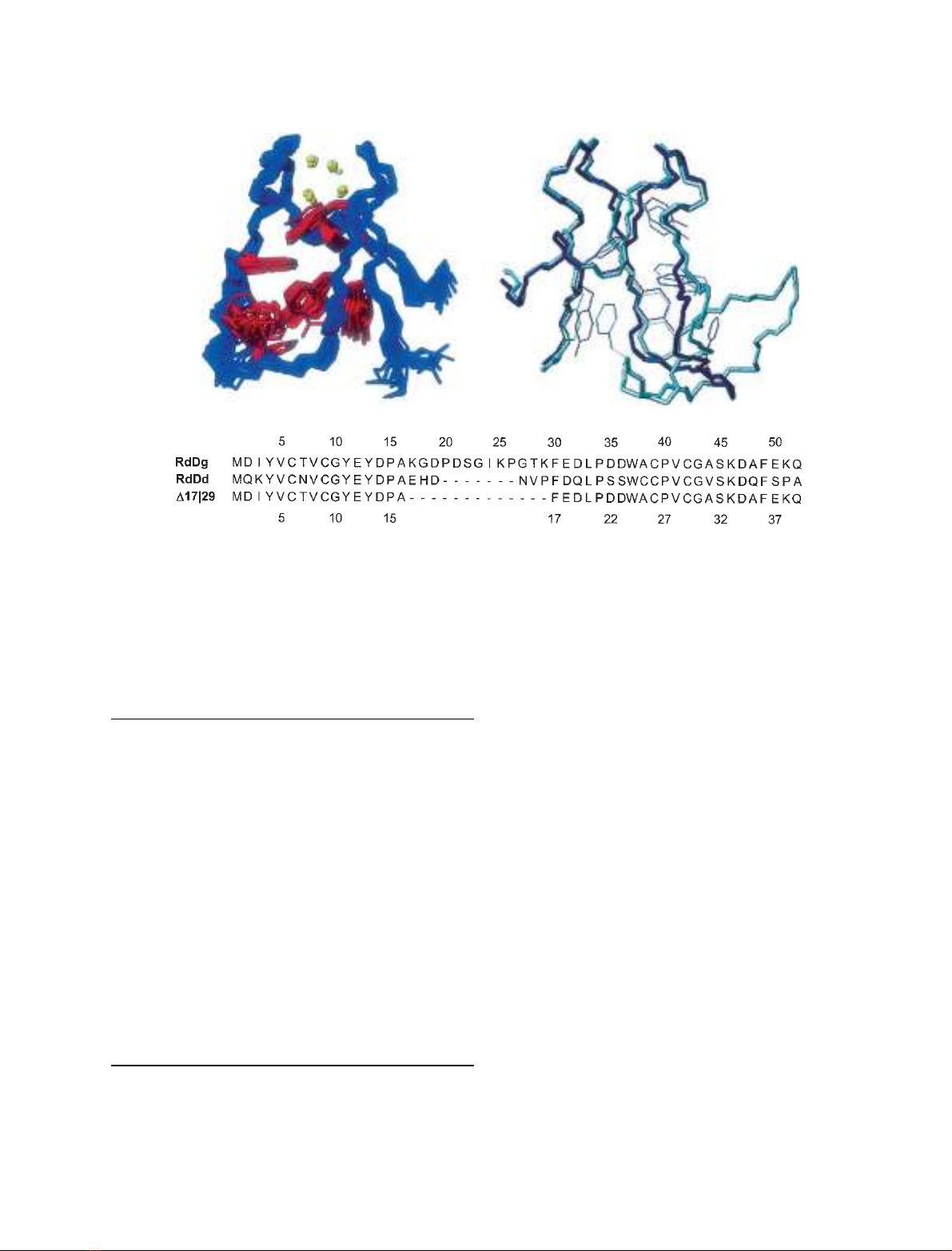
est target functions were selected. A schematic repre-
sentation of the 20 superimposed structures showing
the backbone, aromatic side chains and cysteine sulfur
atoms, is presented in Fig. 2A and a statistical analysis
is given in Table 2. The metal centre conserves both
the geometry and the chirality of the native protein
and is well defined, with the heavy atoms of the four
coordinating cysteines (residues 6, 9, 26 and 29) having
an RMSD < 0.55 A
˚(Fig. 3). Analysis of the secon-
dary structure with molmol v. 2.6 [12] and procheck-
nmr showed the presence of a three-stranded b-sheet
similar to that of the native protein (Fig. 2B). The
Ramachandran plot shows that most of the residues
(94.7%) fall in the most favoured and additionally
allowed regions; however, 5.2% appear in the gener-
ously allowed and one residue (Asp19) appears in the
disallowed region in one of the 20 structures. This resi-
due is located in the residual loop of the mutant and,
if only well-defined regions are considered (Table 2),
no residue appears in the disallowed region. The
deviation is probably a consequence of the large dele-
tion (25% of the residues were deleted) straining the
Table 1. Effect of solute addition on the half-life values (min) for
the thermal denaturation of native rubredoxins and mutants.
Protein No additions
Diglycerol
phosphate 0.1 M
Mannosylglycerate
0.2 M
RdDg
a
96.2 ± 9.4 295.0 ± 7.1 129.6 ± 5.2
D17|29
b
10.5 ± 1.5 16.0 ± 5.0 28.3 ± 0.9
D17|26 14.1 ± 1.4 15.9 ± 2.1 25.2 ± 5.8
D23|29 29.7 ± 3.8 15.1 ± 2.5 45.0 ± 2.1
D2K 77.6 ± 5.5 150.7 ± 4.6
K17E 55.5 ± 4.1 98.7 ± 6.7
V8N 33.5 ± 2.1 36.5 ± 2.1
RdDd
c
30.0 ± 4.0 35.7 ± 4.0
a
The half-life in the presence of 0.2 MKCl is 104 ± 13 min.
b
The
half-life values in the presence of 0.2 MKCl and 0.4 Mtrehalose are
11.2 ± 2.1 min and 19 ± 1.7 min, respectively.
c
Values from Lamo-
sa et al.[7].
Fig. 1. Effect of diglycerol phosphate and mannosylglycerate on
the thermal stability of Desulfovibrio gigas rubredoxin and several
mutants. The half-life values for the thermal denaturation of pro-
teins in the absence of solutes (empty bars), with 0.1 Mdiglycerol
phosphate (solid bars) or with 0.2 Mmannosylglycerate (striped
bars) are depicted.
T. M. Pais et al. Structural determinants of protein stabilization
FEBS Journal 272 (2005) 999–1011 ª2005 FEBS 1001
backbone to accommodate the conserved structural
features.
Overall, the structure of the mutant retains the main
features of the native structure with the obvious excep-
tion of the loop region. The RMSD between the back-
bones of the mean structures for the native and
mutant rubredoxins is 2.24 A
˚. However, if residues 16–
22 (sequence numbering of the mutant), which make
up the shortened loop region in the mutant, are exclu-
ded, the deviation decreases to 0.82 A
˚, showing that
this large deletion left the remaining structure virtually
unaltered (Fig. 2B). The optimal hydrogen bond net-
work was calculated for each of the 20 structures
and it is also similar to that displayed by the native
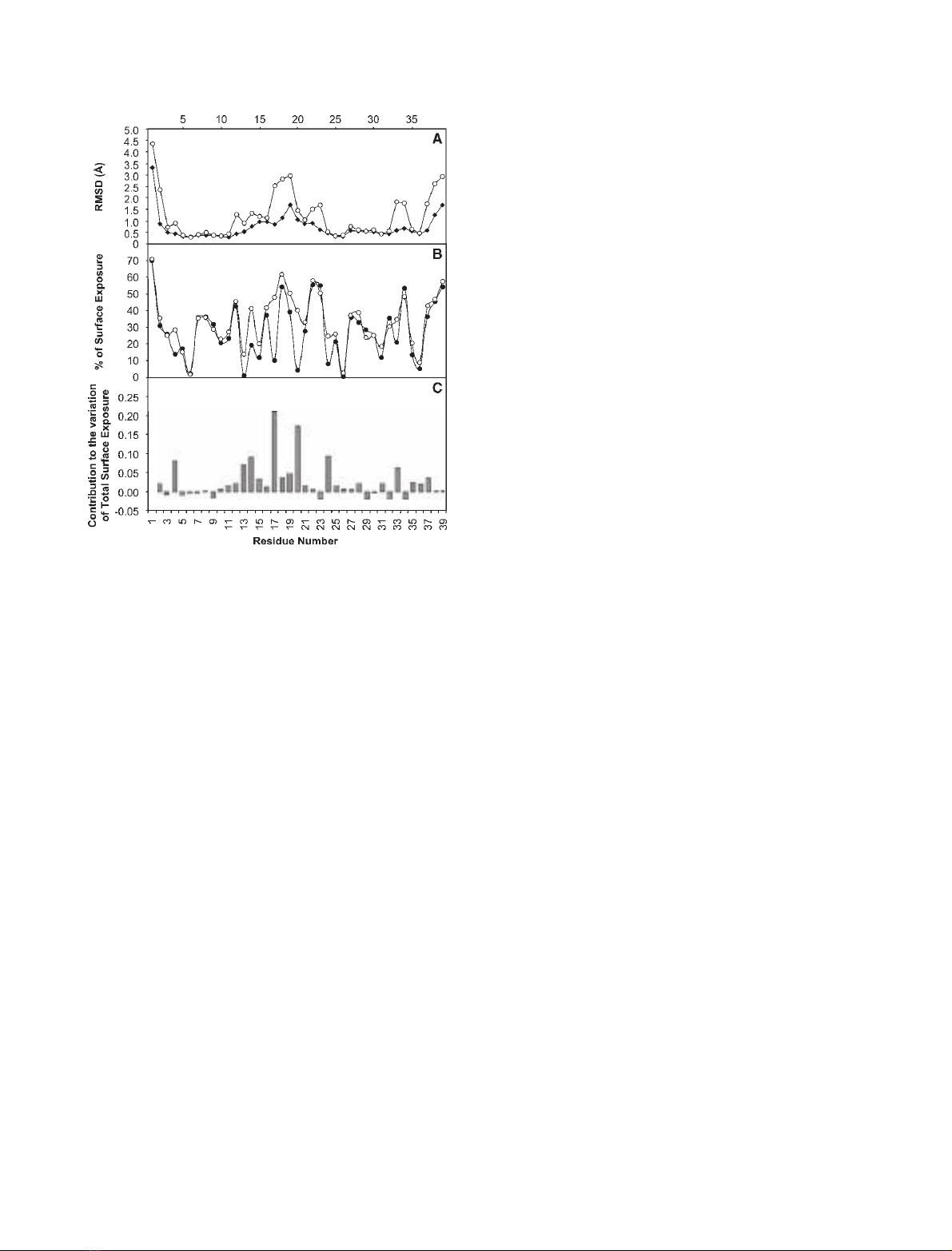
protein [13]. However, the average exposure to water
increased, especially in segment 16–23, with values
over 40% observed for some of these residues (Fig. 3).
In particular, the exposure of the residues that com-
prise the lower part (relative to the orientation depic-
ted in Fig. 2) of the hydrophobic core of the native
protein, namely, Y4, Y13, F17, L20 and W24 (num-
bering according to the mutant) increased substan-
tially.
28 8
9
24
13
4C
N
17
C
N
AB
C
Fig. 2. NMR structure of mutant D17|29 and comparison with the structure of native Desulfovibrio gigas rubredoxin. The 20 best NMR struc-
tures calculated for the mutant are depicted on the left. Only the backbone (blue), aromatic heavy side chains (red) and cysteine sulfur atoms
(yellow) are shown. The right-hand panel shows the superimposition of the native (light blue) and mutant (dark blue) backbone and aromatic
side chains using all residues except those in the loop region. The sequence alignment of the mutant D17|29 and the native rubredoxins
from D. gigas (RdDg) and D. desulfuricans (RdDd) is shown in the lower panel; the upper numbering refers to the RdDg sequence while the
lower refers to the mutant.
Table 2. Restraint violations and quality analysis for the rubredoxin
D17|29 mutant structure.
DYANA target function
Average total (A
˚) 0.21 ± 0.021
Function Range 0.16–0.23
Violated Constraints
Consistent violations (> 0.2 A
˚)0
van der Waals violations (> 0.2 A
˚)0
Precision (A
˚)
Mean global backbone RMSD 0.98 ± 0.21
Mean global heavy atom RMSD 1.72 ± 0.26
Ramachandran plot (%)
a
Most favoured 58.3 (54.5)
Additionally allowed 36.5 (40.2)
Generously allowed 5.2 (5.2)
Disallowed 0 (0.2)
Nonredundant distance restraints (lower limits)
Intraresidual 109
Sequential (|i-j| ¼1) 102
Medium range (2 ¼|i-j| < 5) 92
Long range (|i-j| > 5) 138
Total redundant and nonredundant 734
a
Residues with S (/)andS(w) < 0.8 were not included for the
Ramachandran plot calculation; the values obtained using all resi-
dues are shown in parentheses.
Structural determinants of protein stabilization T. M. Pais et al.
1002 FEBS Journal 272 (2005) 999–1011 ª2005 FEBS
The structure of mutant D17|29 shows considerable
similarities with RdDd, a protein naturally truncated
in the loop region. In fact, excluding residues 16–22
(sequence numbering of the mutant), the RMSD
between the backbones of the X-ray model of RdDd
and the mutant rubredoxin is only 1.22 A
˚. However, if
the residues corresponding to the residual loop region
of mutant D17|29 are included, the RMSD between its
backbone and that of RdDd increases to 2.64 A
˚. The
most striking difference between mutant D17|29 and
RdDd is the absence of a histidine residue in the
mutant protein and the 6.2 A
˚shift of Phe17 (30 in the
RdDd sequence).
Dependence of chemical shifts on solute
concentration
Chemical shifts are sensitive probes of protein confor-
mation. Thus, in an effort to explore possible struc-
tural alterations that solutes might induce in the
protein, or preferential interactions with specific pro-
tein loci, the chemical shifts of all assigned protons in
D17|29 zinc rubredoxin were measured in the presence
of different solute concentrations. Variation of NH
amide chemical shifts along the protein backbone dem-
onstrated an intriguing pattern common to DGP and
MG (Fig. 4) with the major shift variations occurring
in the truncated loop region. This led to two hypothe-
ses: either the action of both solutes upon the structure
was very similar, or the observed shifts were a conse-
quence of increasing ionic strength. To distinguish
between these two hypotheses, KCl (a charged solute,
without significant effect on the half-life values) and
trehalose (a solute that retarded iron loss and with no
charge) were also used to measure chemical shift varia-
tions (Fig. 4). KCl presented a pattern that is very sim-
ilar to those observed with the other charged solutes,
while the effect of trehalose, also concentrated in the
same region, presented much smaller shifts that tended
to be of the opposite sign. However, when the effect of
ionic strength is removed, the shifts observed in the
presence of DGP and MG become comparable in size
with those displayed with trehalose (Fig. 5).
Significant shifts were observed for other types of
protons on solute addition. However, these were not
monotonic with solute concentration and showed no
obvious pattern. Correlation between experimental
chemical shifts and several parameters, such as solvent
exposure, RMSD, secondary shift and temperature
coefficients were also analysed but no obvious good
correlation was found (not shown).
Temperature dependence of amide chemical
shifts
In general, the proton chemical shifts depended line-
arly on temperature, with the smallest coefficients
observed in the metal binding loops (Fig. 6). The bind-
ing sequences X-Cys-X-X-Cys-Gly-X (X ¼variable
amino acid) are largely conserved among rubredoxins,
and comprise residues Val5 to Tyr11, and Ala25 to
Ala31 in the D17|29 mutant. We found a reasonably
good correlation between the existence of hydrogen
bonds and amide protons with small absolute tem-
perature dependence (values more positive than
)4.5 ppbÆK
)1
have been proposed to be a reliable indi-
cator of H-bonding especially if combined with slow
exchange rates) [14]. In the structure of mutant D17|29,
among the residues with high probability of being
involved in H-bonds according to analysis with the
whatif software, 79% have temperature coefficients
above )4.5 ppbÆK
)1
(Fig. 6A). The presence of DGP
in the sample produced a generally small, but consis-
tent increase in the temperature coefficients of amide
protons, with the exception of Lys33 and Ala35, which
Fig. 3. Average RMSD values for each residue and respective aver-
age surface exposure. (A) RMSD values for the backbone (r)and
heavy atoms (s). (B) Percentage of average surface exposure per
residue of mutant A17|29 (s) and wild-type D. gigas rubredoxin
(RdDg) (d). (C) The contribution of each residue to the variation of
the total surface exposure of the mutant protein with respect to
the wild-type RdDg.
T. M. Pais et al. Structural determinants of protein stabilization
FEBS Journal 272 (2005) 999–1011 ª2005 FEBS 1003















