
Role of the hinge peptide and the intersubunit interface
in the swapping of N-termini in dimeric bovine seminal RNase
Carmine Ercole
1
, Francesca Avitabile
1
, Pompea Del Vecchio
1
, Orlando Crescenzi
1
, Teodorico Tancredi
2
and Delia Picone
1
1
Dipartimento di Chimica, Universita
`di Napoli Federico II, Italy;
2
Istituto Chimica Biomolecolare del CNR, Napoli, Italy
Bovine seminal ribonuclease (BS-RNase) is the only known
dimeric enzyme characterized by an equilibrium between
two different 3D structures: MxM, with exchange (or
swapping) of the N-terminal 1–20 residues, and M¼M,
without exchange. As a consequence, the hinge region 16–22
has a different tertiary structure in the two forms. In the
native protein, the equilibrium ratio between MxM and
M¼M is about 7 : 3. Kinetic analysis of the swapping pro-
cess for a recombinant sample shows that it folds mainly in
the M¼M form, then undergoes interconversion into the
MxM form, reaching the same 7 : 3 equilibrium ratio. To
investigate the role of the regions that are most affected
structurally by the swapping, we expressed variant proteins
by replacing two crucial residues with the corresponding
ones from RNase A: Pro19, within the hinge peptide, and
Leu28, located at the interface between subunits. We
compared the structural properties of the monomeric forms
of P19A-BS-RNase, L28Q-BS-RNase and P19A/L28Q-BS-
RNase variants with those of the parent protein, and
investigated the exchange kinetics of the corresponding
dimers. The P19A mutation slightly increases the thermal
stability of the monomer, but it does not alter the swapping
tendency of the dimer. In contrast, the L28Q mutation sig-
nificantly affects both the dimerization and swapping pro-
cesses but not the thermal stability of the monomer. Overall,
these results suggest that the structural determinants that
control the exchange of N-terminal arms in BS-RNase may
not be located within the hinge peptide, and point to a crucial
role of the interface residues.
Keywords: bovine seminal ribonuclease; domain swapping;
proline; ribonuclease A; site-directed mutagenesis.
Bovine seminal ribonuclease (BS-RNase), the only dimeric
protein in the pancreatic-type ribonuclease family, is
characterized in solution by an equilibrium between two
different structures [1]: in the form dubbed MxM, the
N-terminal arms are exchanged, or swapped, between the
two identical subunits, whereas in the form indicated as
M¼M no swapping occurs. In the native protein, the
equilibrium ratio between MxM and M¼M is about 7 : 3.
The two identical subunits are linked through two disulfide
bridges between Cys31 and 32 of one subunit with Cys32¢
and 31¢, respectively, of the partner subunit. Each subunit
has 83% of the amino-acid sequence identical with that of
bovine pancreatic RNase A. In particular, both enzymes
exhibit active sites constituted by identical amino-acid
residues in the same sequence position. Beside ribonuclease
activity, BS-RNase is endowed with several additional
biological activities, such as allostery [2], cytotoxicity toward
malignant cells [3], immunosuppression and antispermato-
genesis [4]. Domain swapping in BS-RNase was found to be
determinant for all of these activities, which may suggest a
physiological role for this structural peculiarity.
A folded and stable monomeric derivative of BS-RNase
can be obtained by selective reduction of the dimeric protein
with a moderate excess of dithiothreitol, and stabilized by
either alkylation of the exposed thiol groups [5] or reaction
with glutathione [6]. All monomeric derivatives of
BS-RNase are catalytically more active than the native
dimeric enzyme, but they do not exhibit any allosteric
property and have no detectable specialbiological action [7].
In a recent paper, we reported an NMR characterization
of the N67D variant of monomeric BS-RNase [8], hence-
forth called mBS. The mutation avoids sample heterogen-
eity arising from the spontaneous deamidation of Asn67 [9],
but it does not affect enzymatic activity. Comparison of the
solution structures, as well as specific NMR relaxation
experiments, indicated that the hinge region 16–22 is much
more flexible in mBS than in RNase A. However, this
region shows the greatest sequence difference from RN-
ase A: GNSPSSS in BS-RNase vs. STSAASS in RNase A.
As a consequence of its flexibility, the structure of this
segment is not well defined in the solution structure of mBS
(Fig. 1A). Moreover, owing to extensive overlap of diag-
nostic signals, we could not unequivocally assign trans
isomerism to Pro19. Mutagenic studies have shown that
Pro19 and Leu28, which in BS-RNase makes a hydrophobic
contact at the interface between the two subunits (Fig. 1B),
are two crucial residues in inducing dimerization and
swapping N-terminal arms in RNase A variants [10,11].
As the first step of a study aimed to investigate, through a
Correspondence to D. Picone, Dipartimento di Chimica, Universita
`
di Napoli Federico II, Via Cintia, 80126, Napoli, Italy.
Fax: + 39 081 674409, Tel.: + 39 081 674406,
E-mail: picone@chemistry.unina.it
Abbreviations: BS-RNase, bovine seminal ribonuclease; mBS, mono-
meric N67D BS-RNase; RNase A, bovine pancreatic ribonuclease;
DVS, divinyl sulfone.
(Received 1 August 2003, revised 2 October 2003,
accepted 7 October 2003)
Eur. J. Biochem. 270, 4729–4735 (2003) FEBS 2003 doi:10.1046/j.1432-1033.2003.03872.x

systematic mutagenic approach, the role played by the hinge
and interface regions in the swapping process, we prepared
BS-RNase variants by replacing Pro19 and Leu28 with the
corresponding residues from RNase A. Here we report a
characterization of monomeric P19A, L28Q and P19A/
L28Q variants of mBS carried out by 2D NMR, CD and
differential scanning microcalorimetry, and an investigation
of the kinetics of swapping of all variant dimers in
comparison with that of the parent protein.
Materials and methods
Construction of mBS mutants
Site-directed mutagenesis was performed by a megaprimer
PCR method [12] to produce the mutants coding for P19A-
mBS, L28Q-mBS and P19A/L28Q-mBS, starting from the
pET-22b(+) plasmid cDNA coding for the wild-type
enzyme which already carries the N67D mutation, to avoid
sample heterogeneity by spontaneous deamidation at the
Asn67 site [8].
PCR amplification was performed with an Eppendorf
Mastercycler amplifier. The forward flanking primer
sequence used in these experiments, 5¢-GAGTGCGGCC
GCAAGCTTGGGCTG-3¢, had an estimated T
m
of 82 C.
The reverse flanking primer sequence, 5¢-ATATACA
TATGAAAGAAAG-3¢, had a calculated T
m
of 42 C.
The mutagenic primers for each variant are: P19A (5¢-AGA
GCTGCTAGCAGAGTTG-3¢) and L28Q (5¢-CACAT
CATCCTGGTTGCAA-3¢) (nucleotides that represent
mutations are underlined). For the mutant P19A/L28Q-
mBS the mutagenic primer L28Q was used starting from the
pET-22b(+) plasmid cDNA coding for the mutant P19A-
mBS. The amplified, mutated genes were separated, excised,
and purified from the agarose gel followed by cloning into
pET-22b(+) between the HindIII and NdeIsites.
Insertion of the correct mutations was confirmed by
DNA sequencing.
Recovery of proteins
All the proteins were expressed in Escherichia coli and
purified in monomeric form, with Cys31 and 32 linked to
two glutathione molecules, as described previously [13].
Monomers with Cys31 and 32 in the reduced form were
obtained by selective reduction of the mixed disulfide
bridges with a 5 : 1 molar excess of dithiothreitol for 20 min
at room temperature in 0.1
M
Tris/acetate buffer, pH 8.4.
The samples were either carboxyamidomethylated with
iodoacetamide [5], to obtain the monomeric proteins used
for CD and microcalorimetric analysis, or dialyzed against
0.1
M
Tris/acetate, pH 8.4, for 20 h at 4 C, to obtain
dimers. The last step of the purification procedure was
always a gel filtration on Sephadex G-75 to separate
monomers from dimers. All dimerization steps were
performed at 4 C.
Recombinant RNase A was obtained and purified as
described previously [8].
Protein homogeneity was verified by SDS/PAGE
and MALDI-TOF MS, registered at the Sezione di
Fig. 1. Ribbon representation of the solution
structure of mBS-RNase (A), as derived from
heteronuclear NMR data (pdb accession code
1WQW), and the X-ray structure of the MxM
form of BS-RNase (B; pdb accession code
1BSR). Pro19 and Leu28 are highlighted. The
figure was drawn with
MOLMOL
software [25].
4730 C. Ercole et al.(Eur. J. Biochem. 270)FEBS 2003
Spettrometria di Massa of the CIMCF, Universita
`degli
Studi di Napoli Federico II. Protein concentration was
measured by UV spectrophotometry.
Kinetics of interconversion of dimeric forms
To follow the interconversion kinetics, dimer samples were
incubated at 37 C. At given times, aliquots were with-
drawn, the interchain disulfide bridges were selectively
reduced as described above [1], and the mixture was
chromatographed on an analytical Superdex 75 HR 10/30
column. The amount of MxM and M¼M was evaluated
quantitatively by integrating the peaks of dimer and
monomer, respectively.
Assessing the extent of the N-terminal swap
at equilibrium
Cross-linking experiments were performed using divinyl
sulfone (DVS) as a 10% solution in ethanol. The protein
(20 lg) in sodium acetate buffer (100 m
M
,pH5,100lL)
and DVS (1 lL of the 10% solution) was incubated at
30 C [11]; this is 1000-fold excess of sulfone over each
subunit of the protein. Aliquots were withdrawn over a
period of 96 h, quenched with 2-mercaptoethanol (final
concentration 200 m
M
), incubated for 15–30 min at room
temperature, and loaded on gels for reducing SDS/PAGE.
The ratio of monomer to cross-linked dimer was estimated
qualitatively by Coomassie blue staining.
NMR
NMR measurements were performed on Bruker DRX400
and DRX500 spectrometers. All spectra were collected
using the standard Bruker pulse sequence library. Protein
concentration was 2 m
M
in 95% H
2
O/5% D
2
O, pH 5.65.
CD
The CD spectra were recorded with a Jasco J-715 spectro-
polarimeter equipped with a Peltier-type temperature con-
trol system (model PTC-348WI). The instrument was
calibrated with an aqueous solution of
D
-10-camphorsulf-
onic acid at 290 nm [14]. Molar ellipticity per mean residue,
[h] in degreesÆcm
2
Ædmol
)1
, was calculated from the equation
[h]¼h
obs
mrw/10lC,whereh
obs
is the ellipticity measured in
degrees, mrw is the mean residue molecular mass (117 Da
[5]), Cis the protein concentration in gÆL
)1
,andlis the
optical path length of the cell in cm. A 0.1-cm path length
cell and a protein concentration of 0.3 mgÆmL
)1
in 10 m
M
sodium acetate buffer, pH 5.0, were used. CD spectra were
recorded at 25 C with a time constant of 16 s, a 2-nm band
width, and a scan rate of 5 nmÆmin
)1
; they were signal-
averaged over at least five scans, and baseline-corrected by
subtracting the buffer spectrum. Thermal unfolding curves
were recorded in the temperature scan mode at 222 nm
from 25 Cupto85C with a scan rate of 0.5 KÆmin
)1
.
Scanning calorimetry
Calorimetric measurements were performed on a second-
generation Setaram Micro-DSC. A scanning rate of
0.5 CÆmin
)1
was chosen for all experiments. The raw data
were converted into an apparent molar heat capacity taking
into account the instrument calibration curve and the
buffer–buffer scanning curve, and by dividing each data
point by the scan rate and the protein molar concentration
in the sample cell. Finally, the excess molar heat capacity
function, <DCp>, was obtained after baseline subtraction,
assuming as reference the heat capacity of the native state
[15].
Results
Recombinant mBS and its P19A, L28Q and P19A/L28Q
variants (P19A-mBS, L28Q-mBS and P19A/L28Q-mBS,
respectively), all with Cys31 and 32 linked to two glutathi-
one molecules, were obtained in pure form with a yield of
about 15 mgÆper L culture. Each of these variants retains a
catalytic activity against yeast RNA comparable with that
of parent mBS, indicating that a native conformation is
present. A further indication of the similarity of their global
fold to that of the parent protein is provided by the 1D
1
H-NMR spectra (data not shown), which display all the
characteristic signals in almost identical positions. The
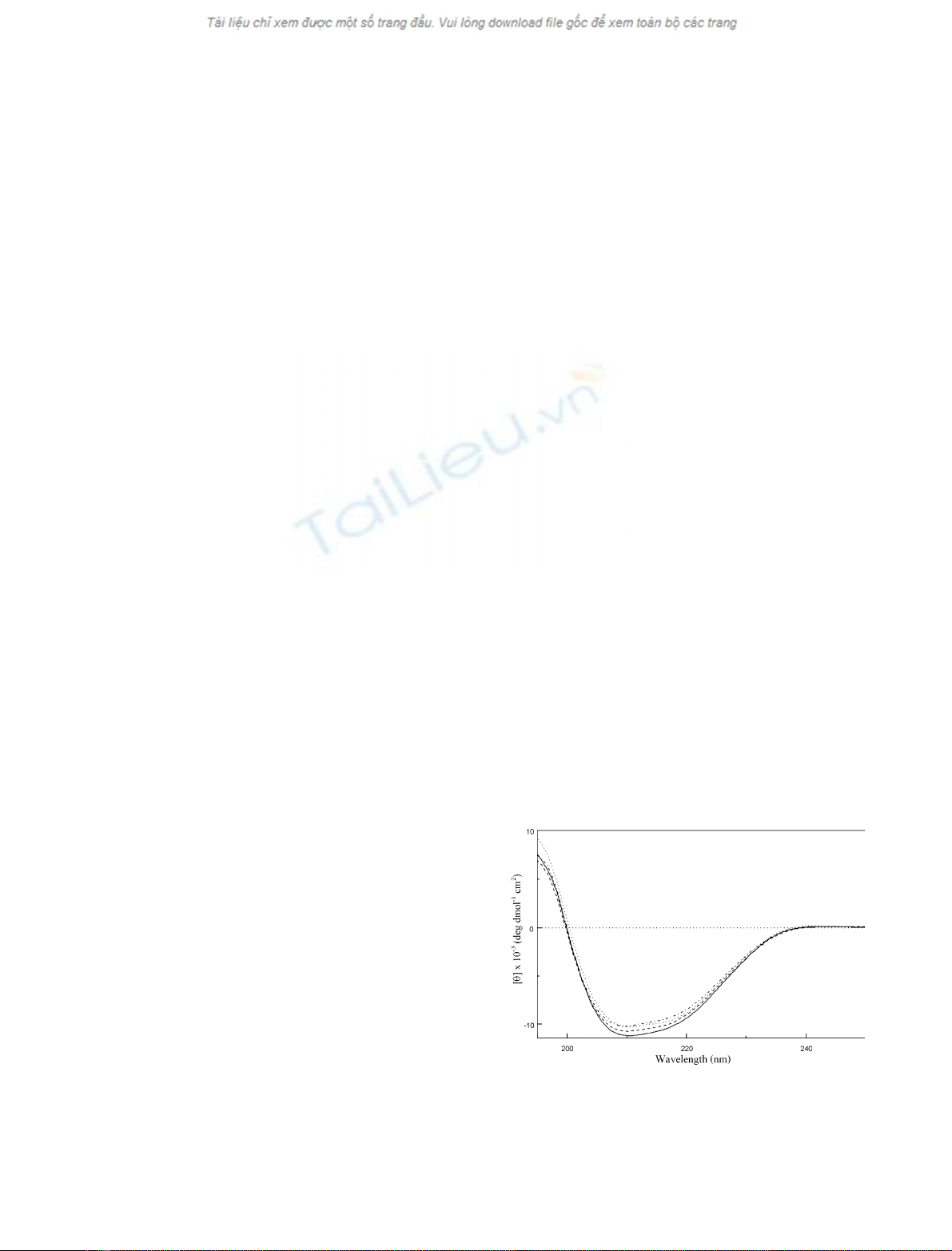
similarity was confirmed by CD measurements (Fig. 2).
The estimation of secondary-structure content, performed
by the neural network-based procedure implemented in the
program
K
2
D
[16,17], yielded very similar values for all the
protein samples (28% a-helix, 36% b-sheet and 40%
random coil); these values are also in good agreement with
the secondary structure derived from the NMR structure of
mBS [8].
To allow a more accurate evaluation of the effect of single
point mutations on the solution structure of monomeric
derivatives, we analysed the 2D NMR spectra of the
different variants of mBS. Figure 3 shows the expanded
regions of TOCSY spectra of L28Q-mBS (panel L28Q),
P19A/L28Q-mBS (panel PALQ) and P19A-mBS (panel
P19A), in comparison with the same region of the parent
mBS (panel mBS). The new signal at 8.40–1.40 p.p.m.,
which appears in the spectra of P19A-mBS and
Fig. 2. Far-UV spectra of mBS-RNase (solid curve), P19A-mBS-RNase
(dashed curve), L28Q-mBS-RNase (dotted/dashed curve) and P19A/
L28Q-mBS-RNase (dotted curve) in 10 m
M
sodium acetate buffer,
pH 5.0, 25 C. The horizontal dotted line indicates the zero value of the
ellipticity.
FEBS 2003 Structural properties of BS-RNase variants (Eur. J. Biochem. 270) 4731


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)