Vietnam Journal of Biotechnology 21(4): 745-757, 2023
745
METAGENOMIC CHARACTERIZATION OF ARCHAEAL AND
BACTERIAL COMMUNITIES ASSOCIATED WITH CORAL, SEDIMENT,
AND SEAWATER IN A CORAL REEF ECOSYSTEM OF PHU QUOC
ISLAND, VIETNAM
Nguyen Thi Phuong Thao1,2,†, Vu Minh Ngoc3,4,†, Pham Van Tra3,4, Bui Van Ngoc2,3,*
1Institute of Biological and Food Technology, Hanoi Open University, B101 Nguyen Hien
Street, Hai Ba Trung District, Hanoi, Vietnam
2Graduate University of Science and Technology, Vietnam Academy of Science and
Technology, 18 Hoang Quoc Viet Road, Cau Giay District, Hanoi, Vietnam
3Institute of Biotechnology, Vietnam Academy of Science and Technology, 18 Hoang Quoc
Viet Road, Cau Giay District, Hanoi, Vietnam
4Hanoi University of Science, Vietnam National University, 334 Nguyen Trai Road, Thanh
Xuan District, Hanoi, Vietnam
†These authors have contributed equally to this work
*To whom correspondence should be addressed. E-mail: bui@ibt.ac.vn
Received: 07.11.2023
Accepted: 27.12.2023
SUMMARY
Recent advancements in metagenomics, particularly in the studies of conservative 16S rRNA
sequences, have significantly accelerated our understanding of the relationship between corals
and their associated microbial communities. While bacteria are known to be closely linked with
corals, there is limited understanding of the connections between archaea and corals. Unlike
previous 16S rRNA studies conducted in similar tropical coral reef ecosystems, we analyzed
both the archaeal and bacterial communities associated with Acropora sp. and Lobophyllia sp.
corals, as well as the surrounding sediment and water columns in Phu Quoc Island, Kien Giang
Province, Vietnam. The data collected were sequenced using the 16S rRNA sequencing method
and further analyzed using bioinformatics tools in the R programming language, employing
DADA2 and phyloseq pipelines. We examined the compositions and diversity of bacteria and
archaea in coral, sediment, and water column samples to establish potential connections between
these two domains. The results revealed that archaea constituted a small percentage of all
samples, averaging 3.18% in coral mucus and reaching an average of 7.49% in sediment
samples. Among the most abundant archaeal taxa were Crenarchaeota and Nanoarchaeota,
alongside bacterial taxa Gammabacteria, Cyanobacteria, and Desulfobacteria, which are
associated with important metabolic processes within coral hosts. Alpha and beta diversity
analyses confirmed the highest archaeal diversity in sediment samples and the distinct existence
of microbial communities in each biotope. These findings complement our knowledge of
archaea’s presence and potential roles in the coral-associated microbiome.
Keywords: 16S rRNA, metagenomics, archaea, bacteria

Nguyen Thi Phuong Thao et al.
746
INTRODUCTION
Coral reefs are regarded as one of the
most diverse and complex marine
ecosystems, encompassing a multitude of
coral species (Wagner et al., 2020; Zhang et
al., 2021a, 2021b). Millions of people
depend on coral reefs for the development of
industries such as fisheries, tourism, food,
and medicine (Eddy et al., 2021).
Additionally, coral reefs provide a favorable
habitat for numerous microbial species and
play a crucial role in coastal protection
against erosion (Elliff, Silva, 2017).
However, coral reefs face the phenomenon of
coral bleaching, which reduces their
coverage (Gardner et al., 2003; Bruno, Selig,
2007; Silverstein et al., 2015; Hughes et al.,
2018; Harrison et al., 2019). This
phenomenon is primarily attributed to
climate change-induced ocean warming,
which negatively impacts the symbiotic
relationship between corals and their
associated microorganisms (Ritchie, 2006;
Rosenberg et al., 2009; Lesser, 2011;
MacKnight et al., 2021). This highlights the
urgent need for research on the diversity and
interactions of microbial communities in
coral reef ecosystems.
Coral provides three habitat niches for
bacteria: the surface mucus layer, coral tissue
(including the gastrovascular cavity), and the
calcium carbonate skeleton, each hosting
distinct bacterial communities (Bourne,
Munn, 2005; Koren, Rosenberg, 2006).
Initially, research on coral-associated
microbes focused on the surface mucus layer,
utilizing traditional culture-based methods.
These studies revealed that this layer harbors
diverse and abundant beneficial bacteria,
including nitrogen-fixing bacteria and chitin-
degrading bacteria (Ducklow, Mitchell,
1979; Williams et al., 1987; Shashar et al.,
1994; Lesser et al., 2004). However, due to
the limitations of culture-based approaches,
only a small fraction of environmental
microbes (0.001-0.01%) were isolated
(Kogure et al., 1979).
Recently, the continuous development of
metagenomics has provided a broader
overview of microbial communities in the
environment, particularly enabling the study
of environment-independent microbial
communities using 16S rRNA sequences
(Pootakham et al., 2017). Next-generation
sequencing technology (NGS) has been
utilized to sequence and analyze the 16S
rRNA gene of microbial communities in
various coral species, aiming to assess
microbial diversity and identify prevalent
symbiotic taxa in different coral species
(Meenatchi et al., 2020). Overall, while there
is extensive research on bacteria, studies on
archaea are relatively scarce. Attempts to
characterize 16S rRNA archaea have been
proven challenging due to their low
abundance in coral mucus, with many studies
struggling to identify them and often
detecting them at very low levels (<0.5%)
(Kellogg, 2004; Littman et al., 2011; Frade
et al., 2016). Nonetheless, archaea are
present in coral reef ecosystems without
forming direct relationships with corals due
to their interaction with anaerobic bacteria in
potential anaerobic microenvironments
within coral mucus layers (Wegley et al.,
2004). Investigations of archaea’s unique
metabolic capabilities, particularly their
participation in anaerobic metabolism, may
expand our understanding of the scope of
interactions between marine hosts and their
associated microbial communities.
To gain a deeper understanding of
microbiota in coral reef ecosystems, we
conducted a metagenomic analysis of the
diversity and composition of archaea and
bacteria in samples collected from Phu Quoc

Vietnam Journal of Biotechnology 21(4): 745-757, 2023
747
Island, Kien Giang, Vietnam. Samples were
acquired from coral reefs inhabited by
Acropora sp. and Lobophyllia sp., as well as
from the sediment layer beneath the seabed
and the water column above. By
implementing established bioinformatics
pipelines and employing appropriate
statistical analysis methods, we aimed to
elucidate the characteristics of archaea and
bacteria within the coral-associated
microbiomes.
MATERIALS AND METHODS
The 16S rRNA data in this study was
provided by the Department of
Bioinformatics, Institute of Biotechnology,
Vietnam Academy of Science and
Technology (VAST). Six samples were
collected from healthy coral branches on Phu
Quoc Island, Vietnam (9°55′20.6″N
104°01′16.4″E) in May 2020. These colonies
hosted Acropora millepora, Acropora
formosa, and Lobophyllia sp. (also known as
brain corals). Additionally, five samples each
from the sediment and the water column
surrounding the coral colonies were gathered
simultaneously. A total of 16 samples were
utilized for DNA extraction and purification.
Subsequently, polymerase chain reaction
(PCR) amplification of the microbial 16S
rRNA gene was carried out using the
following primer set: 5’-
CAGCMGCCGCGGTAA-3’ (forward) and
5’-GTGCTCCCCCGCCAATTCCT -3’
(reverse). The amplified libraries underwent
sequencing using the Illumina MiSeq short
read-sequencing system (San Diego, USA).
The forward and reverse reads were 250
bases in length and were provided in FastQ
format.
Processing and downstream analysis of
16S rRNA sequencing reads were performed
in R using RStudio version 4.3.1. We
implemented the Bioconductor’s DADA2
pipeline (Callahan et al., 2016) for quality
control, trimming, and filtering of sequences.
The first 10 bases containing primers and
adapters were trimmed from all reads.
Additionally, reads containing ambiguous
bases or having a quality score lower than 20
were excluded from each sample.
Subsequently, forward reads were truncated
at position 240, while reverse reads were
truncated at position 210. Following this,
forward and reverse reads were merged and
clustered into amplicon sequencing variants
(ASVs) using a similarity threshold of 97%.
Taxonomic classification was then assigned
to each ASV using the SILVA database
version 138.1 (https://www.arb-silva.de/).
The average relative abundance of taxa was
expressed as mean ± standard deviation.
Further computational analysis, statistical
tests, and visualization were performed using
the R’s phyloseq, vegan, and ggplot2
packages.
Alpha and beta diversity analyses were
also conducted to evaluate the microbial
diversity of the collected samples. Alpha
diversity metrics assess the taxonomic
richness within individual communities or
samples, while beta diversity examines the
diversity across distinct communities
(Andermann et al., 2022). Alpha analysis
indices including Observed, Chao1, and
Shannon were computed, followed by testing
the differences between indices of different
sample types using analysis of variance
(ANOVA). For beta analysis, we employed
principal coordinates analysis (PCoA),
known as multidimensional scaling, which is
a technique used to quantify and visualize the
distance between observations or samples in
a low-dimensional space (Zuur et al., 2007).
Bray-Curtis’s dissimilarity method was

Nguyen Thi Phuong Thao et al.
748
applied to measure sample distances (Bray,
Curtis, 1957). Subsequently, analysis of
similarities (ANOSIM) was used to assess
statistically significant differences between
groups of microbial communities with 1000
permutations.
RESULTS AND DISCUSSION
Taxonomic composition of archaea and
bacteria
A total of 721,652 sequencing reads were
produced from 16 samples. After filtering out
low-quality reads, trimming, denoising, and
removal of chimera, 301,399 reads remained
and were clustered into 4,101 ASVs using
the standard DADA2 pipeline. Using the
SILVA database version 138.1 for taxonomic
classification, taxa belonging to archaea and
bacteria were identified in all samples with
varying compositions (Table 1). The
dominant component of all sample types was
bacteria with an abundance higher than 90%
in all samples. The microbiome associated
with A. formosa and A. millepora exhibited a
relatively low occurrence of archaea,
(<2.0%) while brain corals contained a
considerably high composition of archaea
(6.65% to 7.39%). Sediment samples
contained the highest abundance of archaea,
averaging 7.49% per sample. The water
column also harbored certain archaea,
ranging from 3.57% to 8.10% in relative
abundance.
Table 1. Relative abundance of archaea and bacteria in corals, sediment, and water column.
Location
Sample
Archaea (%)
Bacteria (%)
Corals
AF1
0.51
99.49
AF2
0.83
99.17
AM1
1.75
98.25
AM2
1.97
98.03
BR1
7.39
92.61
BR2
6.65
93.35
Sediment
SD1
9.15
90.85
SD2
8.76
91.24
SD3
6.87
93.13
SD4
6.36
93.64
SD5
6.33
93.67
Water column
WC1
3.57
96.43
WC2
6.25
93.75
WC3
6.02
93.98
WC4
5.47
94.53
WC5
8.10
91.90
Vietnam Journal of Biotechnology 21(4): 745-757, 2023
749
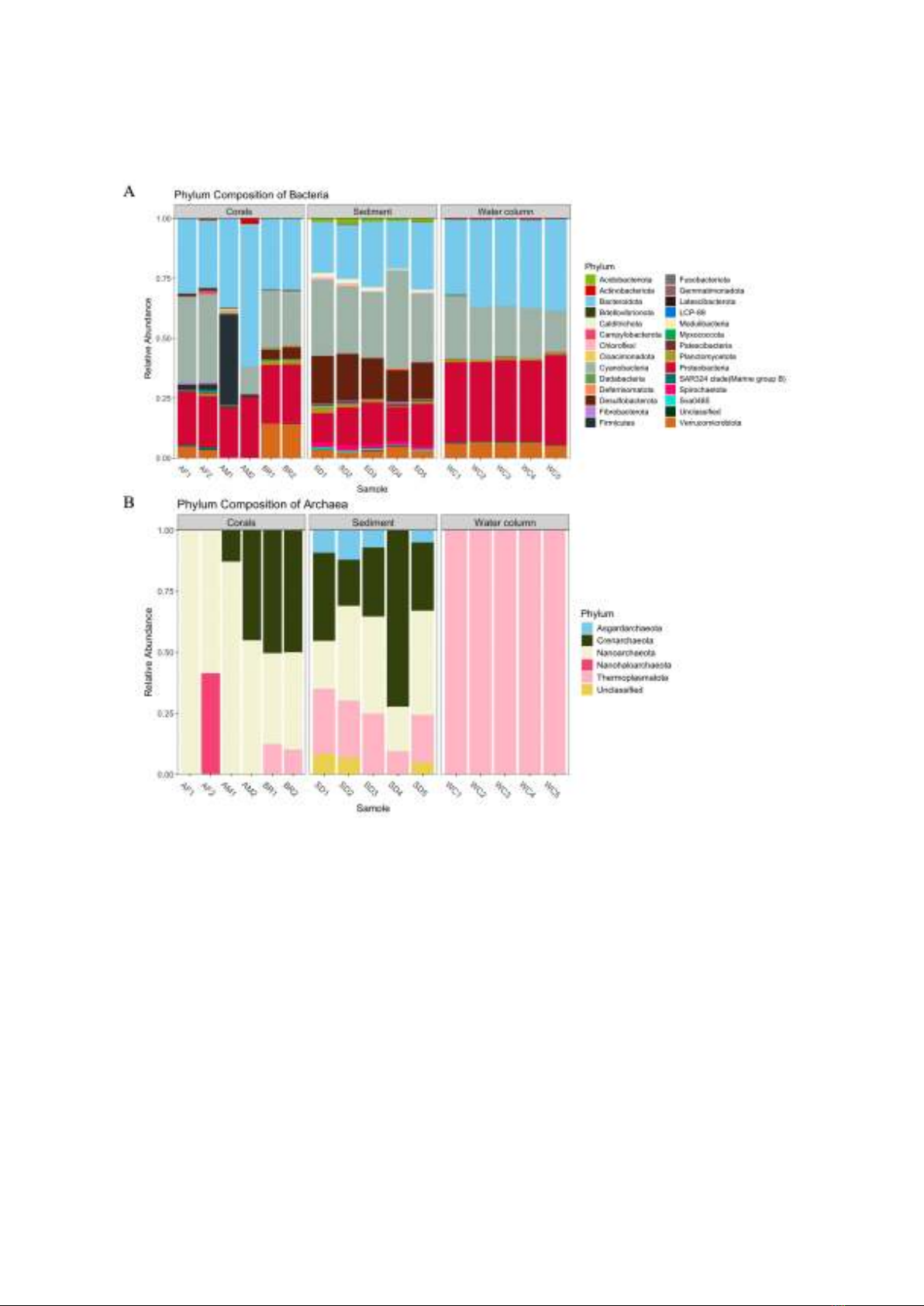
Figure 1. Composition at phylum level of bacteria (A) and archaea (B) in coral, sediment, and water
column samples (AF: A. formosa, AM: A. millepora, BR: brain corals). The abundance threshold was
set to 0.2% to filter out all low-abundant phyla.
Using the SILVA database, we identified
a total of 5 phyla, 7 classes, 6 orders, 4
families, and 2 genera of archaea in all
samples. However, no archaeal species were
characterized. Regarding the bacterial
community, 27 phyla, 44 classes, 95 orders,
112 families, 167 genera, and 12 species
were found. Figure 1 depicts the phylum-
level composition of both archaeal and
bacterial communities.
Archaea were considerably more diverse
in sediment compared to other specimens,
hosting four out of five phyla detected in all
samples. These include Asgardarchaeota,
Crenarchaeota, Nanoarchaeota, and
Thermoplasmatota, averaging 8.4±3.0%,
36.7±20.8%, 31.8±11.8%, and 20.8±6.8% in
abundance respectively, with Nanoarchaeota
and Crenarchaeota consistently making up
more than 56% of archaea across all











![Giáo trình Vi sinh vật học môi trường Phần 1: [Thêm thông tin chi tiết nếu có để tối ưu SEO]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20251015/khanhchi0906/135x160/45461768548101.jpg)





![Bài giảng Sinh học đại cương: Sinh thái học [mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20250812/oursky02/135x160/99371768295754.jpg)



![Đề cương ôn tập cuối kì môn Sinh học tế bào [Năm học mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260106/hoang52006/135x160/1251767755234.jpg)

![Cẩm Nang An Toàn Sinh Học Phòng Xét Nghiệm (Ấn Bản 4) [Mới Nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20251225/tangtuy08/135x160/61761766722917.jpg)