36
TỔNG QUAN VỀ MÃ DNA TRONG VIỆC PHÂN LOẠI
VÀ XÂY DỰNG CÂY PHÁT SINH PHÂN TỬ
Bùi Thị Kim Lý1
1. Khoa Y Dược, Trường Đại học Thủ Dầu Một, liên hệ email: lybtk@tdmu.edu.vn
TÓM TẮT
Nhu cầu định danh sinh vật đã và đang dần trở nên quan trọng không chỉ trong các nghiên
cứu về sinh thái, bảo tồn đa dạng sinh học mà còn ứng dụng trong đời sống con người như phân
loại vi sinh vật hay truy xuất nguồn gốc nông, thuỷ sản. Sự phát triển nhanh chóng của khoa học
công nghệ đã cho phép ứng dụng nhiều kỹ thuật hiện đại trong phân tích trình tự DNA và mối liên
hệ của chúng. Việc xác định khoảng cách di truyền giữa các trình tự có ý nghĩa trong việc xây
dựng cây phát sinh phân tử, từ đó mô tả được mối quan hệ tiến hoá giữa các trình tự mang đi phân
tích. Bài tổng hợp này cung cấp các khái niệm chung nhất về mã DNA, khoảng cách di truyền của
chúng và việc xây dựng mối liên hệ tiến hoá giữa các trình tự, nhằm định hướng trong ứng dụng
nghiên cứu đa dạng sinh học, khẳng định loài mới, hay truy xuất nguồn gốc động, thực vật.
Từ khóa: DNA barcoding, định danh, phylogeny, khoảng cách di truyền
1. MÃ DNA TRONG PHÂN LOẠI, TRUY XUẤT NGUỒN GỐC THỰC VẬT
Sự phát triển của khoa học công nghệ hiện đại ngày nay đã mở ra các hướng tiếp cận mới
trong việc ứng dụng các kỹ thuật sinh học phân tử vào việc phân loại và định danh sinh vật.
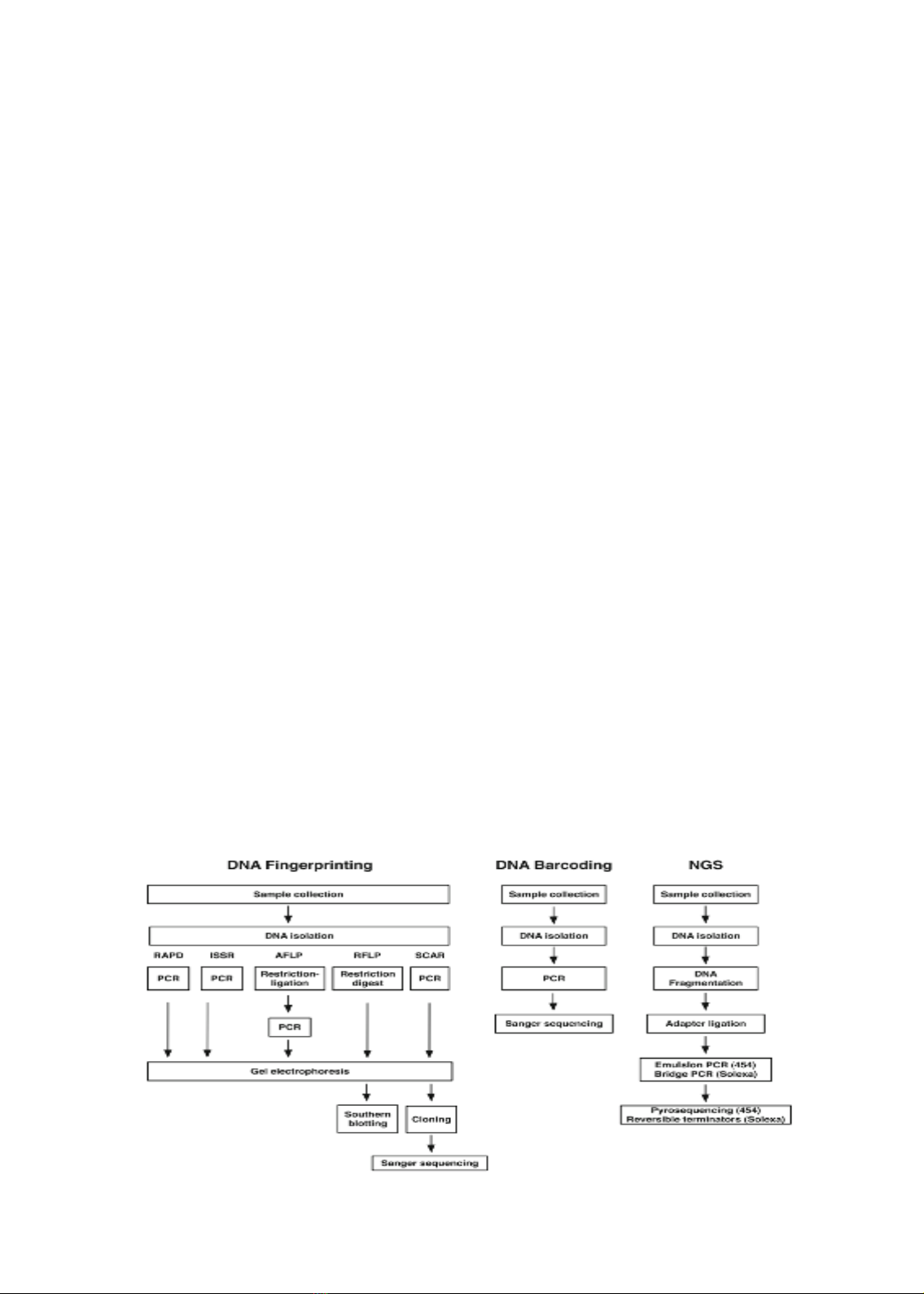
DNA fingerprinting là một trong những kỹ thuật được ứng dụng đầu tiên và được xem là công
cụ độc lập trong việc điều tra pháp y, nghiên cứu và nhiều lĩnh vực khác (Sucher và nnk, 2012).
Sau sự xuất hiện của kỹ thuật khuyếch đại PCR và giải trình tự (Sequencing) thì khái niệm về
DNA Barcoding đã ra đời và được ứng dụng như một phương tiện hỗ trợ đầy triển vọng trong
việc phân loại các loài và cá thể (Kress và nnk, 2012; Sucher và nnk, 2012). Hiện nay bên cạnh
DNA Barcoding và DNA Fingerprinting, kỹ thuật giải trình tự gen thế hệ mới (NGS) cũng được
ứng dụng nhiều trong việc phân loại, và nghiên cứu sinh thái các loài (Sucher và nnk, 2012).
Hình 1. Các định hướng tiếp cận đi đôi với kỹ thuật phân tích trong quá trình định danh sinh vật

37
DNA barcoding là khái niệm được mô tả lần đầu năm 2003 nhằm chỉ các vùng trình tự
gen ngắn thuộc các vùng tiêu chuẩn trong bộ gen, được dùng như công cụ để xác định và phân
biệt các loài với nhau (Sucher và nnk, 2012). Kể từ lúc ra đời, DNA barcoding trở thành kỹ
thuật được xây dựng và phát triển mạnh mẽ nhằm thay thế DNA fingerprints. Năm 2004, hiệp
hội Barcode for life (CBOL) ra đời nhằm xây dựng các định hướng, tiêu chuẩn trong hệ thống
nghiên cứu và quản lý barcode của sinh vật. Năm 2007, dự án tiêu chuẩn DNA barcode của
thực vật trên cạn được CBOL công bố (Sucher và nnk, 2012). Nghiên cứu DNA barcoding được
xem là mang nhiều triển vọng trong nhiều ngành nghiên cứu như: nghiên cứu tiến hóa thông
qua tần số thay đổi trình tự của loài thông qua thời gian; nghiên cứu đa dạng sinh học thông qua
việc phân biệt, định danh các cá thể và dự đoán các loài mới phát hiện; chỉ dấu theo dõi, nghiên
cứu các đối tượng đặc biệt; hoặc để truy xuất nguồn gốc, và nhiều ứng dụng khác (Kress, 2017).
Để một DNA barcoding được tạo ra và áp dụng thì đòi hỏi phải có hai bước cơ bản: bước 1 là
xây dựng thư viện mã vạch gồm tập hợp tất cả trình tự các loài liên quan trên một hay một
nhóm gen đánh dấu (marker) mục tiêu xác định, các nhóm cá thể cung cấp trình tự này phải
được xác định loài cụ thể và có các giấy tờ chứng nhận đi kèm, đây sẽ là các hồ sơ cần thiết đi
suốt cùng barcode được cấp; bước 2 là nhận diện cá thể mới thông qua việc giải trình tự vùng
marker thuộc mã vạch ở bước 1 sau đó dùng các thuật toán để ghép nối, gióng cột trình tự của
cá thể mới và thư viện mã vốn có để đưa ra nhận xét và kết luận về sự tương đồng của cá thể
(Kress, 2017). Đây là phương pháp được tiếp cận gần đây, và hiện vẫn còn nhiều tranh cãi trong
việc sử dụng DNA barcoding trong phân loại (Kress, 2017). Sự phân loại và các tiêu chuẩn kèm
theo, phụ thuộc vào tần suất khác biệt của các loài trong cùng một họ, và vùng trình tự được
mang đi làm mã (Bellafronte và nnk, 2013).
Trên động vật, vùng gen cytochrome C oxidase (viết tắt là Cox1 hay CO1) được xem là
vùng gen barcode lõi vì có khả năng phân biệt được rất nhiều loài động vật khác nhau. Tuy
nhiên đối với thực vật, các nghiên cứu cho thấy vùng gen trên các loài thực vật có rất ít biến
đổi và không phù hợp cho phân loại (Sucher và nnk, 2012). Các nghiên cứu sau này cũng cho
thấy khả năng sử dụng DNA barcode trên thực vật là kém hiệu quả hơn nhiều so với động vật.
Chẳng hạn như hệ gen của ty thể ở thực vật vì một số lý do nào đó mà sự phát triển của hệ gen
ty thể ở thực vật lại diễn ra đồng thời với sự di chuyển môi trường sống từ nước lên cạn của
chúng, trong khi các vùng gen ty thể ở động vật lại có tính bảo tồn cao. Bên cạnh đó, hệ genome
ở thực vật chịu nhiều ảnh hưởng của hình thức sinh sản đơn tính hay hữu tính, đồng thời có sự
tương đồng cao giữa các loài hơn so với động vật. Chính vì vậy mà hệ gen lạp thể lại được chú
ý như nguồn trình tự có thể ứng dụng trong DNA barcode, tuy nhiên vẫn gặp nhiều khó khăn
vì tính tương đồng trong hệ gen plasmid và nhiều nhóm thực vật hầu như không có các đặc
trưng, sai khác trong trình tự plasmid. Hiện nay CBOL chỉ công nhận hai vùng gene trên lạp
thể là matK và rbcL như DNA barcode chính cho phân loại thực vật đồng thời đề xuất thêm
một số vùng gen khác là rbcL trên lạp thể, ITS trên genome và hai vùng non-coding atpF-atpH
và trnH-psbA (Sucher và nnk, 2012).
2. KHOẢNG CÁCH DI TRUYỀN GIỮA HAI VÙNG TRÌNH TỰ
Các trình tự marker gene chỉ có ý nghĩa phân biệt khi khoảng cách di truyền nằm trong giới
hạn nhất định. Khoảng cách giữa các mã vạch DNA được xem như khoảng cách di truyền xét
trên một vùng gen marker dùng cho phân biệt loài (Hebert và nnk, 2003). Khoảng cách mã vạch
được ghi nhận từ các loài hay các cụm marker gen khác nhau có sự biến động lớn. Trong điều
kiện lý tưởng, khoảng cách 4% là tỉ lệ sai khác thấp nhất để có thể phân biệt hai loài khác nhau,
tuy nhiên trên các marker gen không đặc hiệu thì con số này có thể lên hơn 10% (Meyer và nnk,
2005). Để giảm thiểu các tác động khách quan gây ảnh hưởng lên việc phân biệt loài, việc xác

38
định khoảng cách di truyền trong nội bộ loài (giữa các thứ, chủng cùng loài) là cần thiết để làm
thước đo khi so sánh loài này với các loài khác (Steinke và nnk, 2009). 10X là khoảng đề xuất để
phân biệt một loài là khác biệt so với quần thể đang tham chiếu (Shen và nnk, 2016).
Hình 2. Một số mô hình đề xuất trong việc tính toán khoảng cách di truyền giữa các trình tự (A);
ví dụ cụ th cho việc lựa chọn mô hình phù hợp cho trình tự phân tích (B)
Để đo lường được khoảng cách giữa hai trình tự, các mô hình đã được đưa ra từ đơn giản
đến phức tạp như đo khoảng cách p (p-distance) bằng cách lấy tỉ lệ nucleotide sai khác trên
tổng số nucleotide mang so sánh (Masatoshi và nnk, 2002), mô hình JC69, mô hình K80, mô
hình HKY85, mô hình TN93 và nhiều mô hình khác. Trong đó, mô hình hai tham số K2P (K80
của Kimura) được xem là mô hình phù hợp trong việc phân tích khoảng cách giữa hai loài gần
nhau (Shen và nnk, 2016). Trong mô hình này, Kimura đã phân biệt riêng sự thay đổi nucleotide
thành hai dạng transitions (thay đổi giữ hai nucleotide có cùng dạng vòng thơm là purine hoạc
pyrimidine) hoặc transversions (thay đổi nucleotide khác dạng vòng thơm) từ đó hình thành hai
tỷ lệ thay đổi tương ứng hai biến số trong mô hình (Kimura, 1981). Hình 2A đề cập đến các mô
hình và tham số tương ứng mà mô hình đó quan tâm trong việc tính toán khoảng cách di truyền.
3. CÂY PHÁT SINH PHÂN TỬ: Ý NGHĨA VÀ SỰ HÌNH THÀNH
3.1. Cây phát sinh phân tử
Thuật ngữ phát sinh phân tử được dùng để mô tả việc ứng dụng các thuật toán trong so
sánh các trình tự nucleotide (hoặc amino acid) từ đó nêu lên mối quan hệ tiến hóa của các trình
tự mang đi so sánh (Brown, 2002). Kết quả các mối liên hệ trong di truyền của các nhóm trình
tự được hình thành và mô tả thông qua dạng đồ thị được gọi là cây phát sinh phân tử (Harrison
và nnk, 2006; Masatoshi và Sudhir, 2002). Cả trình tự polypeptide và nucleotide đều có thể
được sử dụng cho mục tiêu xác định mối liên hệ về di truyền giữa các nhóm mẫu mang đi phân
tích, tuy nhiên vẫn còn nhiều tranh cãi xung quanh tính chính xác trong việc sử dụng hai trình
tự này cho phân tích bởi lẽ có tới 20 ký tự cho amino acid trong khi chỉ có 4 kí tự cho nucleic
acid (Harrison và Langdale, 2006). Cấu trúc cây phát sinh phân tử được cấu tạo từ các nút
(node) và các nhánh (branch), trong khi các nhánh đại diện cho sự ổn định của trình tự thì các
39
nút là vị trí bắt đầu cho một trình tự mới khác biệt (xem xét là loài mới), chiều dài của nhánh
cho biết được khoảng cách khác biệt giữa các nút (Brown, 2002; Yang và nnk, 2012). Cây phát
sinh phân tử có thể được biểu diễn ở dạng có gốc hoặc không có gốc, số lượng cấu trúc có thể
hình thành ở dạng cây có gốc là nhiều hơn so với cây không gốc (Masatoshi và Sudhir, 2002).
Quá trình xây dựng cây phát sinh phân tử có thể tóm gọn bao gồm việc lựa chọn vùng trình tự
phân tích, xếp gióng cột các vùng trình tự, lựa chọn mô hình phân tích và cuối cùng là xây dựng
cây phát sinh phân tử (Ray, 2014).
3.2. Mô hình thiết cập cây phát sinh phân tử
Các phương pháp tính toán được áp dụng để phân tích tất cả các cặp trình tự được đưa
vào và xây dựng cây phát sinh phân tử từ các dữ liệu phân tích được (Yang và Rannala, 2012).
Phương pháp ma trận khoảng cách (bao gồm cả neighbour joining (NJ)) là phương pháp tính
toán khoảng cách di truyền giữ các trình tự dự trên tỉ lệ khác biệt về loại nucleotide (Harrison
và Langdale, 2006; Saitou và nnk, 1987). Đặc điểm của phương pháp là đơn giản và nhanh
chóng dựa trên các mô hình phân tích như mô hình JC69, mô hình HKY85,…(Harrison và
Langdale, 2006; Yang và Rannala, 2012). Phương pháp này thường được sử dụng cho cỡ mẫu
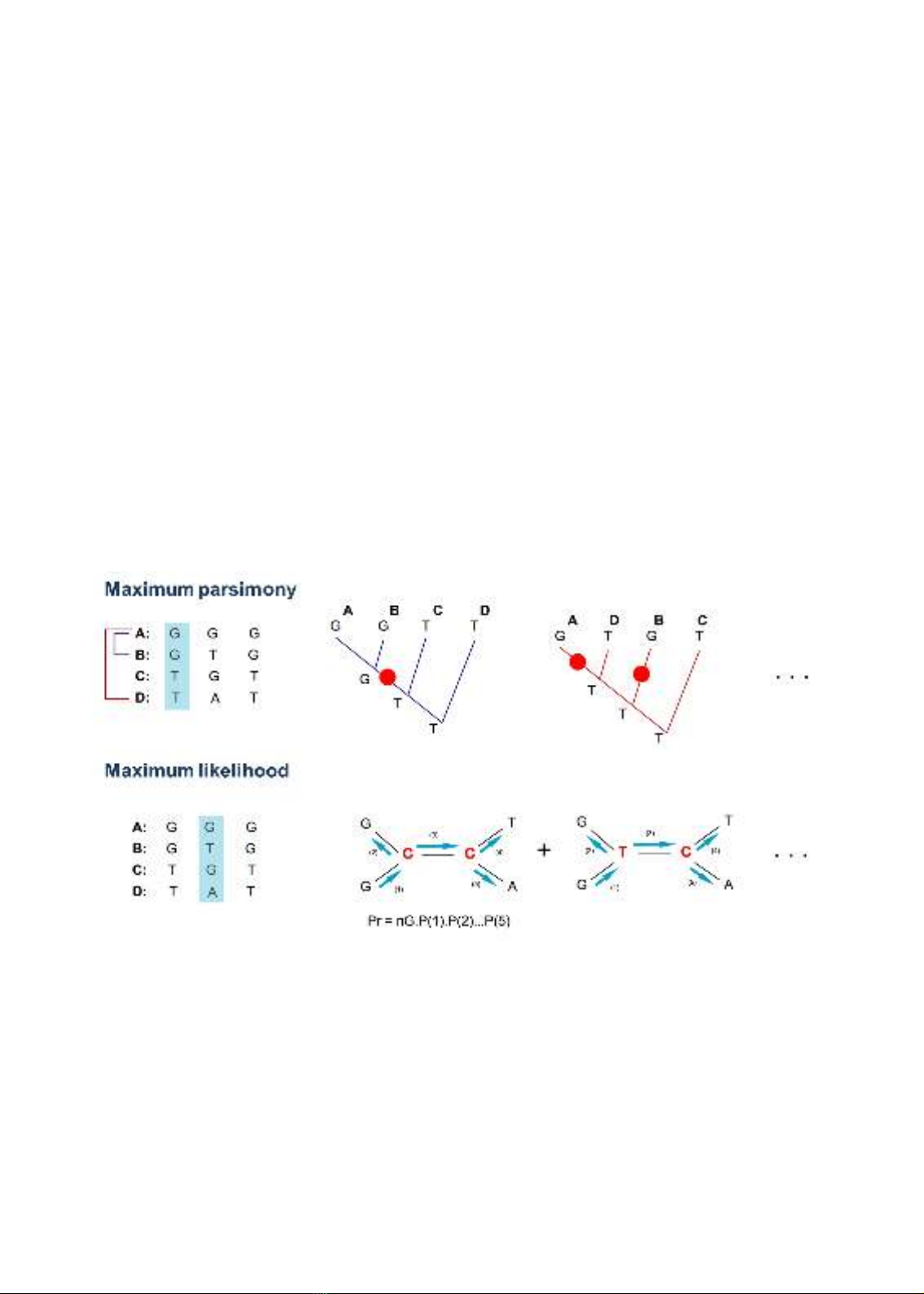
lớn và mức độ tiến hóa của trình tự nhỏ (Yang và Rannala, 2012). Phương pháp maximum
parsimony tối thiểu hóa số lượng các thay đổi trên cây phát sinh hay nói cách khác là các loài
phân tích sẽ được xếp cạnh nhau lần lượt để hình thành cây phát sinh phân tử với ít nhánh nhất.
Đây là phương pháp nhanh chóng và đơn giản trong áp dụng, tuy nhiên có nhiều tranh cãi xung
quanh tính hợp lý và rõ ràng của giả định được đặt ra (Yang và Rannala, 2012).
Hình 3. Sự khác biệt trong hướng tiếp cận của phương pháp maximum parsimony và phương
pháp maximum likelihood
Ngược lại với phương pháp maximum parsimony, phương pháp maximum likelihood
giúp ước lượng các thông số nhằm đưa ra một mô hình cây phát sinh mà có xác xuất dễ xảy ra
nhất từ các dữ liệu cung cấp (Harrison và Langdale, 2006). Phương pháp này cung cấp các giả
định rõ ràng và có kho mô hình phân tích phong phú do đó được ứng dụng rộng rãi trong việc
xây dựng cây phát sinh phân tử, bên cạnh đó việc vận hành phương pháp này thường kèm theo
một số lượng phép tính lớn đòi hỏi có thiết bị phân tích phù hợp, bên cạnh đó việc xác định sai
mô hình phân tích thì các phương pháp thống kê của phương pháp trở nên không hiệu quả
(Yang và Rannala, 2012). Bootstrap là phương pháp hỗ trợ nhằm tăng độ tin cậy cho các điểm

40
nút trong cây phát sinh phân tử. Phương pháp này thực hiện thông qua việc tạo các bộ dữ liệu
giả từ bộ dữ liệu gốc đầu vào và tiến hành phân tích trên các bộ dữ liệu này nhằm xác định ra
cấu trúc cây phát sinh có tỉ lệ xuất hiện cao nhất (Tsagkanos, 2008). Bên cạnh các phương pháp
nêu trên, một số phương pháp, thuật toán khác cũng được ứng dụng, hỗ trợ trong quá trình phân
tích mối liên hệ di truyền giữa các đoạn trình tự như phương pháp Bayesian thông qua ánh xạ
các thông số (Yang và Rannala, 2012).
Việc thực hiện phân tích di truyền trong chứng minh, phân biệt và xây dựng mối liên hệ
loài khá phức tạp, xong nhìn chung cần trải qua các giai đoạn chính bao gồm xác định vùng
trình tự có khoảng cách di truyền thích hợp cho việc phân loại, lựa chọn mô hình phân tích, lựa
chọn phương pháp tiếp cận trong việc xây dựng cây phát sinh phân tử.
TÀI LIỆU THAM KHẢO
1. Bellafronte, E., Mariguela, T., Pereira, L., Oliveira, C., & Moreira-Filho, O. (2013). DNA barcode
of Parodontidae species from the La Plata river basin - applying new data to clarify taxonomic
problems. Neotropical Ichthyology, 11(3), 497-506.
2. Brown, T. A. (2002). Genomes (2nd ed.). Oxford: Wiley-Liss.
3. Harrison, C. J., & Langdale, J. A. (2006). A step by step guide to phylogeny reconstruction. Plant J,
45(4), 561-572.
4. Hebert, P. D., Cywinska, A., Ball, S. L., & deWaard, J. R. (2003). Biological identifications through
DNA barcodes. Proc Biol Sci, 270(1512), 313-321.
5. Kimura, M. (1981). Estimation of evolutionary distances between homologous nucleotide
sequences. Proceedings of the National Academy of Sciences of the United States of America, 78(1),
454-458.
6. Kress, W. J. (2017). Plant DNA barcodes: Applications today and in the future. Journal of
Systematics and Evolution, 55(4), 291-307.
7. Kress, W. J., & Erickson, D. L. (2012). DNA barcodes: methods and protocols (2012/06/12 ed. Vol.
858).
8. Masatoshi, N., & Sudhir, K. (2002). Molecular evolution and phylogenetics (Vol. 25). USA: Oxford
University Press,.
9. Meyer, C. P., & Paulay, G. (2005). DNA Barcoding: Error Rates Based on Comprehensive
Sampling. PLOS Biology, 3(12), e422.
10. Ray, S. (2014). Molecular markers in phylogenetic studies-a review. Journal of Phylogenetics &
Evolutionary Biology, 2(2), 1-9.
11. Saitou, N., & Nei, M. (1987). The neighbor-joining method: a new method for reconstructing
phylogenetic trees. Mol Biol Evol, 4(4), 406-425.
12. Shen, Y., Guan, L., Wang, D., & Gan, X. (2016). DNA barcoding and evaluation of genetic diversity
in Cyprinidae fish in the midstream of the Yangtze River. Ecology and Evolution, 6(9), 2702-2713.
13. Steinke, D., Zemlak, T. S., & Hebert, P. D. N. (2009). Barcoding nemo: DNA-based identifications
for the ornamental fish trade. PLOS ONE, 4(7), e6300.
14. Sucher, N., Hennell, J., & Carles, M. (2012). DNA Fingerprinting, DNA Barcoding, and Next Generation
Sequencing Technology in Plants. Methods in molecular biology (Clifton, N.J.), 862, 13-22.
15. Tsagkanos, A. (2008). The Bootstrap Maximum Likelihood Estimator: the case of logit. Applied
Financial Economics Letters, 4(3), 209-212.
16. Yang, Z., & Rannala, B. (2012). Molecular phylogenetics: principles and practice. Nature Reviews
Genetics, 13(5), 303-314.














![Bài giảng Giáp xác chân mái chèo [mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20250927/lethihongthuy2402@gmail.com/135x160/92891759114976.jpg)



![Tài liệu học tập Chuyên đề tế bào [mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20250906/huutuan0/135x160/56151757299182.jpg)






