
TNU Journal of Science and Technology
230(01): 423 - 431
http://jst.tnu.edu.vn 423 Email: jst@tnu.edu.vn
PHYLOGENETIC RELATIOSHIP OF PANAX SPECIES
IN VIENAM USING MIG-SEQ METHOD
Hoang Thi Binh1, Le Minh Tam2, Pham Quang Tuyen3,
Trinh Ngoc Bon3, Vu Quoc Luan4, Nguyen Van Ngoc1*
1Dalat University, 2Vaccine Company Limited of Dalat Pasteur,
3Silviculture Research Institute - Forest Science Institute of Vietnam,
4Tay Nguyen Institute for Scientific Research - Vietnam Academy of Scientific and Technology
ARTICLE INFO
ABSTRACT
Received:
04/6/2024
This study aim to examine the phylogenetic relationship between Panax
species (Araliaceae) of Vietnam using MIG-seq method based on Next
Generation Sequencing platform. A total of 29 Vietnamese Panax
samples, including five species, two varieties, and one sample of Panax
ginseng as an outgroup, were utilized. The MIG-seq method generated
a dataset of 6821 SNPs, which was used to reconstruct a phylogenetic
tree using maximum likelihood. The tree demonstrated the phylogenetic
relationship between Panax species with high resolution and strongly
supported monophyly through bootstrap values. The results revealed that
the Panax species of Vietnam comprise two main sister groups. The first
group includes P. stipuleanatus and P. bipinnatifidus, while the second
group comprises three species (P. pseudoginseng, Panax sp., P.
vietnamensis) and two varieties (P. vietnamensis var. fuscidicus and P.
vietnamensis var. langbianensis). These research findings provide a
foundation for the application of the MIG-seq method in studying
phylogenetic relationships of plant in Vietnam.
Revised:
17/12/2024
Published:
18/12/2024
KEYWORDS
Araliaceae
Classification
ISSR
Phylogeny
Sequencing
NGHIÊN CỨU MỐI QUAN HỆ PHÁT SINH GIỮA CÁC LOÀI THUỘC CHI
PANAX Ở VIỆT NAM BẰNG PHƯƠNG PHÁP MIG-SEQ
Hoàng Thị Bình1, Lê Minh Tâm2, Phạm Quang Tuyến3,
Trịnh Ngọc Bon3, Vũ Quốc Luận4, Nguyễn Văn Ngọc1*
1Trường Đại học Đà Lạt, 2Công ty TNHH Một thành viên Vắc Xin Pasteur Đà Lạt
3Viện Nghiên cứu Lâm sinh - Viện Khoa học Lâm Nghiệp Việt Nam
4Viện Khoa học Tây Nguyên - Viện Hàn Lâm Khoa học Công nghệ Việt Nam
THÔNG TIN BÀI BÁO
TÓM TẮT
Ngày nhận bài:
04/6/2024
Nghiên cứu này nhằm xây dựng mối quan hệ phát sinh giữa các loài
thuộc chi Panax ở Việt Nam bằng phương pháp MIG-seq trên nền tảng
giải trình tự gene thế hệ mới. Tổng cộng 29 mẫu thuộc các loài Panax
ở Việt Nam và 01 mẫu thuộc loài Panax ginseng (làm nhóm ngoài) đã
được sử dụng. Nghiên cứu đã thu được tổng cộng 6821 điểm đa hình
đơn nucleotide (SNPs) làm chỉ thị phân tử để xây dựng cây phát sinh
chủng loại theo phương pháp maximum likelihood. Cây biểu thị mối
quan hệ phát sinh giữa các loài Panax ở Việt Nam có độ phân giải cao,
với các nhóm đơn hình được hỗ trợ mạnh mẽ bởi các chỉ số bootstrap.
Kết quả nghiên cứu cho thấy, chi Panax ở Việt Nam gồm 2 nhóm có
quan hệ gần gũi, nhóm thứ nhất gồm các loài P. stipuleanatus và P.
bipinnatifidus, nhóm thứ 2 gồm các loài P. pseudoginseng, Panax sp.,
P. vietnamensis và 02 thứ P. vietnamensis var. fuscidicus và P.
vietnamensis var. langbianensis. Kết quả nghiên cứu làm cơ sở cho việc
ứng dụng phương pháp MIG-seq trong nghiên cứu mối quan hệ phát
sinh cho các đối tượng thực vật ở Việt Nam.
Ngày hoàn thiện:
17/12/2024
Ngày đăng:
18/12/2024
TỪ KHÓA
Họ Nhân sâm
Phân loại
ISSR
Phát sinh loài
Giải trình tự
DOI: https://doi.org/10.34238/tnu-jst.10537
* Corresponding author. Email: ngocnv@dlu.edu.vn

TNU Journal of Science and Technology
230(01): 423 - 431
http://jst.tnu.edu.vn 424 Email: jst@tnu.edu.vn
1. Giới thiệu
Chi Panax thuộc họ Nhân sâm (Araliaceae), là một chi thực vật gồm nhiều loài có giá trị dược
liệu cao [1], [2]. Các loài thuộc chi này phân bố chủ yếu ở ba khu vực chính là Đông Bắc Á, Tây
Trung Quốc và Bắc Mỹ [3]. Ở Việt Nam, năm 1969, Grushvitzky và cộng sự (1969) đã ghi nhận
loài Panax bipinnatifidus Seem. trong họ Araliaceae ở miền Bắc Việt Nam [4]. Sau đó, Phạm
Hoàng Hộ đã phát hiện loài P. schinseng Nees var. japonicum Mak. (Sâm Nhật Bản) có phân bố
tại vùng núi Langbiang (năm 1970), và ba loài ở vùng núi phía Bắc và Tây Nguyên Việt Nam (năm
1993) là: P. bipinnatifidus Seem, P. japonica (Nees) Meyer, và P. pseudoginseng Wall (Tam thất
Bắc) [5]. Năm 1985, Sâm Ngọc Linh (P. vietnamensis) được Hà Thị Dung và Grushvitzky phát
hiện và công bố loài mới [6].
Từ năm 2000, nhiều nghiên cứu về thành phần loài chi Panax đã được thực hiện. Theo Nguyễn
Tập (2005), chi này ở Việt Nam có 5 loài, trong đó P. bipinnatifidus Seem, P. stipuleanatus H.T.
Tsai et K.M. Feng (Tam thất hoang) và P. vietnamensis Ha et Grushv. (Sâm Ngọc Linh) là các loài
bản địa, còn P. ginseng (sâm Hàn Quốc) và P. notoginseng (Tam thất) là loài nhập nội [7]. Năm
2013, Phan Kế Long và cộng sự đã ghi nhận một thứ sâm mới P. vietnamensis var. fuscidiscus tại
Mường Tè, Lai Châu và đặt tên địa phương là sâm Lai Châu [8]. Các nghiên cứu sau đó đã phát
hiện thêm nhiều loài và thứ mới, nâng tổng số loài Panax ở Việt Nam lên 6 loài và 3 thứ, bao gồm:
Panax japonicus var. japonicus (Sâm Nhật Bản), P. japonicus var. bipinnatifidus (Sâm Vũ Diệp),
P. stipuleanatus (Tam thất hoang), P. notoginseng (Tam thất), P. pseudoginseng (Tam thất Bắc),
P. vietnamensis var. vietnamensis (Sâm Ngọc Linh), P. vietnamensis var. fuscidiscus (Sâm Lai
Châu), P. vietnamensis var. langbianensis (Sâm Langbian) và P. zingiberensis (Tam thất gừng)
[1]. Trong đó, Panax vietnamensis var. langbianensis (sâm Langbian) được Nông Văn Duy và
cộng sự công bố vào năm 2016 [9].
Mối quan hệ phát sinh chủng loại và lịch sử tiến hóa của chi Panax đã được nghiên cứu bằng
các mã vạch phân tử truyền thống phổ biến như ITS, 18SRNA, trnC-trnD, matK, rbcL hay psbK-
psbL,… [10]-[13]. Ở Việt Nam, việc ứng dụng mã vạch phân tử trong nghiên cứu mối quan hệ phát
sinh các loài thuộc chi Panax gần đây nhận được nhiều quan tâm. Nguyễn Thị Phương Trang và
cộng sự đã sử dụng trình tự gen ITS-rDNA để phân tích lịch sử tiến hoá, mối quan hệ di truyền giữa
sâm Ngọc Linh (P. vietnamensis) và các loài khác thuộc chi Panax, cho thấy P. vietnamensis có
quan hệ gần gũi với P. notoginseng mặc dù có sự sai khác 18 nucleotide [14]. Nhóm nghiên cứu
của Phan Kế Long đã sử dụng trình tự ITS-rDNA và matK để nghiên cứu quan hệ phát sinh giữa
các loài thuộc chi Panax ở Việt Nam, kết quả nghiên cứu đã chỉ ra mối quan hệ gần gũi giữa sâm
Lai Châu và sâm Ngọc Linh [15]. Nguyễn Thị Phương Trang cùng các đồng nghiệp đã sử dụng
các vùng gen rbcL và rpoL để phân biệt các loài sâm Việt Nam [16], trong khi nhóm của Lê Thanh
Hương đã khảo sát tiềm năng của 5 mã vạch DNA ứng dụng trong định loại các loài sâm ở Việt
Nam, kết quả đã xác định ITS và psbA-trnH là chỉ thị đạt hiệu quả cao nhất [17]. Phạm Quang
Tuyến và cộng sự cũng đã dùng trình tự ITS1-5.8S-ITS2 để nghiên cứu đa dạng di truyền, xác định
mối quan hệ tiến hóa của 24 mẫu sâm Lai Châu [18].
Nền tảng giải trình tự gen thế hệ mới (Next Generation Sequencing: NGS) ra đời những năm
2000 đã giúp giảm 50.000 lần chi phí và có thể đọc trên toàn hệ gene (hàng tỉ cặp base) tăng từ 100
đến 1000 lần lưu lượng xử lý so với công nghệ trước đó của dự án giải mã hệ gen người. Dữ liệu
lớn thu được từ các nền tảng giải trình tự gene thế hệ mới (Illumina, IonTorrent, PGM, SOLiD,
454,…) có thể sử dụng trong nghiên cứu lắp ráp, chú giải và lập bản đồ hệ gen (de novo assembly),
phân tích đa hình đơn nucleotide (SNPs/InDel), lắp ráp, chú giải hệ phiên mã (de novo), nghiên
cứu xác định hệ SNPs đặc trưng cho các loài hay các dòng giúp cho công tác định danh, phân loại,
nghiên cứu mối quan hệ phát sinh,…
Kỹ thuật “xác định đặc tính di truyền các đoạn trình tự đơn giản lặp lại (Inter Simple Sequence
Repeats: ISSR) bằng giải trình tự gene” (Multiplexed ISSR genotyping by sequencing: MIG-seq)
được phát triển bởi Suyama và Matsuki năm 2015 dựa trên nền tảng công nghệ giải trình tự gene

TNU Journal of Science and Technology
230(01): 423 - 431
http://jst.tnu.edu.vn 425 Email: jst@tnu.edu.vn
thế hệ mới [19]. MIG-seq đã được Suyama và cộng sự ứng dụng trong phân tích các biến dị di
truyền theo các quy luật Mendel, di truyền quần thể, định danh các dòng, các cá thể và trong phân
tích mối quan hệ phát sinh chủng loại,… [20]. Phương pháp giải trình tự gene MIG-seq cung cấp
các chỉ thị SNPs từ các trình tự đọc tương đối ngắn trải đều trên toàn hệ gene, trái ngược với các
đoạn DNA có bộ gene kéo dài liên tục. MIG-seq tương tự như RAD-seq [21], [22], tuy nhiên MIG-
seq thực hiện một phản ứng PCR, không sử dụng enzyme cắt giới hạn và có thể ứng dụng rộng rãi
cho nhiều loại mẫu hơn là kỹ thuật RAD-seq, kể cả mẫu bảo tàng và hoá thạch, thậm chí với chất
lượng và số lượng DNA rất thấp [19]. Khác với các chỉ thị phân tử truyền thống, các mồi dùng
trong MIG-seq có thể khuếch đại hiệu quả hàng nghìn vùng trên toàn bộ hệ gen kể cả trong điều
kiện chưa có bất kỳ thông tin di truyền gì trước đó, và cho phép phát hiện hàng trăm cho đến hàng
nghìn biến dị di truyền (SNPs, InDel,…).
Ở Việt Nam, việc ứng dụng các phương pháp giải trình tự gene thế hệ mới để nghiên cứu mối
quan hệ phát sinh giữa các loài thuộc chi Panax còn hạn chế, đặc biệt chưa có nghiên cứu nào sử
dụng phương pháp MIG-seq. Trong nghiên cứu này, chúng tôi sử dụng các chỉ thị SNPs có được
từ việc ứng dụng phương pháp MIG-seq để nghiên cứu làm rõ mối quan hệ phát sinh giữa các loài
thuộc chi Panax ở Việt Nam.
2. Vật liệu và phương pháp nghiên cứu
2.1. Vật liệu nghiên cứu
Vật liệu nghiên cứu bao gồm 30 mẫu lá được thu từ các loài thuộc chi Panax ở Việt Nam. Các
mẫu được Phạm Quang Tuyến và Trịnh Ngọc Bon, Viện Khoa học Lâm nghiệp Việt Nam định
danh ban đầu dựa vào các đặc điểm hình thái. Theo đó, vật liệu nghiên cứu bao gồm mẫu của các
loài: sâm Ngọc Linh – P. vietnamensis (P01, P02), tam thất Bắc – P. pseudoginseng (P11, PB1,
PB2, PB3), sâm Vũ Diệp – P. bipinnatifidus (PD1, PD2), sâm Lai Châu - P. vietnamensis var.
fuscidiscus (P03, P04, PV1, PV2), tam thất hoang – P. stipuleanatus (PS1, PS2, PS3, PS4, TT41,
TT41A, TT42, TT42A) và sâm Langbian - P. vietnamensis var. langbianensis (P10). Ngoài ra,
trong nghiên cứu này có sử dụng 8 mẫu chưa được định danh, trong đó 4 mẫu (P06, P07, P08, P09)
được thu từ Lai Châu và 4 mẫu (PT1G, PT2G, PT3G, PT3GA) thu từ Tuyên Quang. Mẫu sâm Hàn
Quốc – P. ginseng (P05) thu tại vườn thực nghiệm khoa Sinh học, trường Đại học Đà Lạt được lựa
chọn làm nhóm đối chứng (out group). Thông tin chi tiết được được mô tả tại Bảng 1.
Bảng 1. Danh sách các mẫu sử dụng trong nghiên cứu
STT
Ký hiệu mẫu
Tên khoa học
Tên thông thường
Địa điểm thu mẫu
1
P01
P. vietnamensis
Sâm Ngọc Linh
Kon Tum
2
P02
P. vietnamensis
Sâm Ngọc Linh
Kon Tum
3
P03
P. vietnamensis var. fuscidicus
Sâm Lai Châu
Tam Đường, Lai Châu
4
P04
P. vietnamensis var. fuscidicus
Sâm Lai Châu
Tam Đường, Lai Châu
5
P05
P. ginseng
Sâm Hàn Quốc
Đại học Đà Lạt
6
P06
Panax sp.
Chưa biết
Tuyên Quang
7
P07
Panax sp.
Chưa biết
Tuyên Quang
8
P08
Panax sp.
Chưa biết
Tuyên Quang
9
P09
Panax sp.
Chưa biết
Tuyên Quang
10
P10
P. vietnamensis var. langbianensis
Sâm Langbian
Lâm Đồng
11
P11
P. pseudoginseng
Tam thất Bắc
Lai Châu
12
PB1
P. pseudoginseng
Tam thất Bắc
Tam Đường, Lai Châu
13
PB2
P. pseudoginseng
Tam thất Bắc
Tam Đường, Lai Châu
14
PB3
P. pseudoginseng
Tam thất Bắc
Tam Đường, Lai Châu
15
PD1
P. bipinnatifidus
Sâm Vũ Diệp
Tam Đường, Lai Châu
16
PD2
P. bipinnatifidus
Sâm Vũ Diệp
Tam Đường, Lai Châu
17
PS1
P. stipuleanatus
Tam thất hoang
Sapa, Lào Cai
18
PS2
P. stipuleanatus
Tam thất hoang
Sapa, Lào Cai
19
PS3
P. stipuleanatus
Tam thất hoang
Sapa, Lào Cai

TNU Journal of Science and Technology
230(01): 423 - 431
http://jst.tnu.edu.vn 426 Email: jst@tnu.edu.vn
STT
Ký hiệu mẫu
Tên khoa học
Tên thông thường
Địa điểm thu mẫu
20
PS4
P. stipuleanatus
Tam thất hoang
Sapa, Lào Cai
21
PT1G
Panax sp.
Chưa biết
Tuyên Quang
22
PT2G
Panax sp.
Chưa biết
Tuyên Quang
23
PT3G
Panax sp.
Chưa biết
Tuyên Quang
24
PT3GA
Panax sp.
Chưa biết
Tuyên Quang
25
PV1
P. vietnamensis var. fuscidicus
Sâm Lai Châu
Tam Đường, Lai Châu
26
PV2
P. vietnamensis var. fuscidicus
Sâm Lai Châu
Tam Đường, Lai Châu
27
TT41
P. stipuleanatus
Tam thất hoang
Sapa, Lào Cai
28
TT41A
P. stipuleanatus
Tam thất hoang
Sapa, Lào Cai
29
TT42
P. stipuleanatus
Tam thất hoang
Sapa, Lào Cai
30
TT42A
P. stipuleanatus
Tam thất hoang
Sapa, Lào Cai
2.2. Phương pháp nghiên cứu
2.2.1. Phương pháp tách chiết DNA
DNA tổng số được tách chiết từ các mẫu lá đã được sấy khô trong túi silicagel bằng phương
pháp CTAB [23] với sự điều chỉnh nhỏ theo mô tả của Toyama và cộng sự [24]. DNA sau khi được
tách sẽ được kiểm tra nồng độ và độ tinh sạch bằng máy NanoDrop (Hoa Kỳ) và được lưu trữ ở
nhiệt độ -40oC để dùng cho các phân tích phân tử tiếp theo.
2.2.2. Phương pháp giải trình tự gene thế hệ tiếp theo (MIG-seq)
DNA tổng số được pha loãng ở nồng độ 10 pM/μl làm khuôn mẫu để khuếch đại hàng nghìn
chuỗi trình tự ngắn của các vùng Inter-simple sequence repeats (ISSR) với bộ mồi PCR tiêu chuẩn
theo Suyama và Matsuki [19], [25]. Mồi xuôi và mồi ngược của phản ứng PCR thứ nhất có chiều
dài 31 nucleotide, bao gồm 14 nucleotide trình tự đuôi, 3 nucleotide mỏ neo, 12 nucleotide vùng
SSR, cuối cùng là 2 nucleotide mỏ neo thứ hai ở đầu 3’. Sự khác biệt giữa mồi xuôi và mồi ngược
chỉ ở trong trình tự đuôi của chúng. Nhiệt độ bắt cặp thấp (48oC) được sử dụng để tăng khả năng
khuếch đại vùng ISSR hơn, bao gồm cả một số bắt cặp không hoàn toàn khớp [19], [20].
Phản ứng PCR đầu tiên được thực hiện nhằm khuếch đại các vùng ISSR từ bộ gene DNA bằng
sự kết hợp PCR với đoạn mồi đuôi ISSR. Theo đó, thành phần của phản ứng PCR thứ nhất bao
gồm: 1 μl DNA mẫu, 0,2 μM của mỗi mồi PCR thứ nhất, 3,5 μl dung dịch đệm PCR (Multiplex
PCR Assay Kit Ver.2, Takara Bio, Kusatsu, Japan), 0,035 μl hỗn hợp enzyme PCR (Multiplex
PCR Assay Kit Ver.2, Takara Bio). Điều kiện thực hiện phản ứng PCR thứ nhất như sau: Bước
kích hoạt ban đầu ở 94°C trong 1 phút, 25 chu kỳ khuếch đại (mỗi chu kỳ khuếch đại bao gồm:
Biến tính ở 94°C trong 30 giây, bắt cặp ở 48°C trong 1 phút, kéo dài ở 72°C trong 1 phút), bước ủ
cuối cùng ở 72°C trong 10 phút. Sản phẩm PCR được kiểm tra bằng hệ thống điện di vi mạch
(MultiNA; Shimadzu, Kyoto, Nhật Bản) với thuốc thử DNA-2500 (Shimadzu).
Mỗi sản phẩm của phản ứng PCR thứ nhất sẽ được pha loãng xuống 50 lần bằng nước cất và
sau đó sử dụng làm mẫu cho phản ứng PCR thứ hai (phản ứng PCR gắn đuôi). Bước này cho phép
thêm các trình tự bổ sung để gắn các vùng của tế bào Flow cell trên máy giải trình tự Illumina và
các chỉ dấu đối với từng mẫu cho các sản phẩm của phản ứng PCR thứ nhất, sử dụng cặp mồi xuôi
và mồi ngược đã được chỉ dấu (index) riêng cho từng mẫu. Trình tự mồi xuôi: 5'-
AATGATACGGCGACCACCGAGATCTACACXXXXXACACTCTTTCCCTACACGACGCT
CTTCCGATCTCTG-3', trình tự mồi ngược: 5'-
CAAGCAGAAGACGGCATACGAGATxxxxxxxxxGTGACTGGAGTTCAGACGTGTGCTCT
TCCGATCTGAC-3' (trong đó "XXXXX" và "xxxxxxxxx’" biểu thị chỉ dấu gồm 5 và 9 base tương
ứng với mồi xuôi và mồi ngược) [19], [20].
Thành phần phản ứng PCR thứ 2 gồm: 2,5 μl sản phẩm PCR thứ nhất đã pha loãng, 2,4 μl dung
dịch đệm PrimeSTAR GXL 5X (Takara Bio), 0,96 μM dNTP mixture, 0,24 μl enzym DNA
polymerase PrimeSTAR GXL (Takara Bio), 1,2 μl mỗi loại mồi xuôi và mồi ngược, 3,5 μl nước
cất. Phản ứng PCR thứ 2 được chạy theo 20 chu kỳ khuếch đại. Mỗi chu kỳ bao gồm: Biến tính ở
TNU Journal of Science and Technology
230(01): 423 - 431
http://jst.tnu.edu.vn 427 Email: jst@tnu.edu.vn
98°C trong 10 giây, bắt cặp ở 54°C trong 15 giây và kéo dài ở 68°C trong 30 giây. Sau đó, 3 μl
mỗi sản phẩm của phản ứng PCR thứ 2 sẽ được cho vào một thư viện hỗn hợp. Hỗn hợp này sau
đó được tinh sạch và các phân đoạn với kích thước từ 350–800 bp sẽ được chọn bởi một hệ thống
chọn lọc kích thước Pipin Prep DNA (Sage Science, Beverly, MA, USA). Nồng độ cuối cùng được
đo bằng phản ứng định lượng PCR, thư viện mẫu với nồng độ khoảng 10 pM sẽ được dùng cho
việc giải trình tự gene trên thiết bị Illumina MiSeq Sequencer (Illumina, San Diego, CA, USA) sử
dụng bộ Kit MiSeq Reagent Kit v3 (150 cycle, Illumina).
Để đảm bảo thu được số lượng và chất lượng trình tự đọc (read) lớn nhất, nhóm nghiên cứu đã
thực hiện chạy 2 lần MIG-seq cho tất cả các mẫu nghiên cứu. Dữ liệu thu được từ 2 lần chạy sẽ
được tổ hợp để có được bộ dữ liệu tối ưu thực hiện các phân tích tiếp theo.
2.2.3. Chọn lọc các điểm đa hình đơn nucleotide (SNPs)
Dữ liệu thu được bằng phương pháp MIG-seq sẽ được kiểm soát chất lượng bằng phần mềm
Trimmomatic phiên bản 0.39 [26]. Sau đó, chương trình Population trong phần mềm Stacks [27]
được sử dụng để gọi các điểm đa hình đơn nucleotide theo các thông số được thiết lập như mô tả
trong các nghiên cứu trước đó [28], [29]. Dữ liệu SNPs thu được sẽ được sử dụng để xây dựng cây
phát sinh chủng loại.
2.2.4. Phương pháp phân tích phát sinh chủng loại
Phần mềm RAxML ver. 8.2 [30] được sử dụng để xây dựng cây phát sinh chủng loại dựa vào
dữ liệu SNPs bằng phương pháp Maximum likelihood và mô hình tiến hoá GTR + G theo khuyến
nghị của jMmodelTest 2.1.1 [31]. Hình thái cây phát sinh được khẳng định bằng việc thực hiện
phương pháp bootstrap lặp lại 1000 lần.
3. Kết quả và bàn luận
3.1. Kết quả tách chiết DNA và giải trình tự gen bằng phương pháp MIG-seq
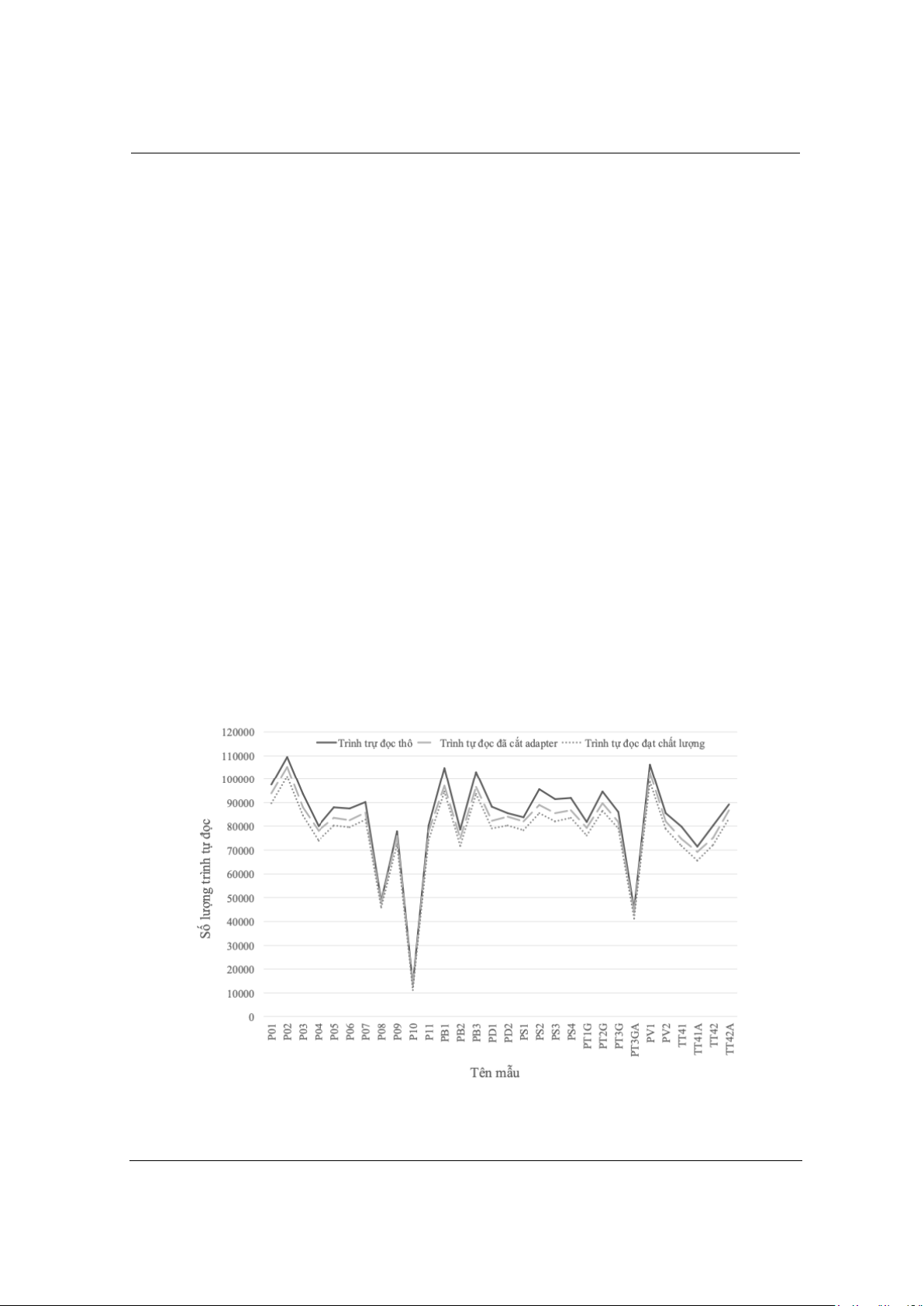
Tất cả 30 mẫu Panax được tách chiết và giải trình tự thành công bằng phương pháp MIG-seq. Theo
hình 1, tất cả các mẫu có số lượng trình tự đọc thô (đường solid line), số lượng trình tự đọc sau khi cắt
adapter (dashed line) và số lượng trình tự đọc đạt chất lượng (dotted line) đạt trên 10000 read.
Hình 1. Tổng số lượng trình tự đọc thu được sau hai lần chạy MIG-seq
Kết quả tổng hợp sau 2 lần chạy MIG-seq thu được mẫu có số lượng trình tự đọc thấp nhất
(11122 read) là P10 (P. vietnamensis var. Langbianensis), mẫu có số lượng trình tự đọc cao nhất


















![Giáo trình Đất trồng phân bón (Nghề Bảo vệ thực vật CĐ/TC) - Trường Cao đẳng Gia Lai [Mới Nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260224/hoacattuong2026/135x160/9591771994804.jpg)
![Giáo trình Kỹ thuật canh tác cây lương thực (Nghề Bảo vệ thực vật - CĐ/TC) - Trường Cao đẳng Gia Lai [Mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260224/hoacattuong2026/135x160/12471771994805.jpg)




![Giáo trình Công nghệ sau thu hoạch (Nghề Bảo vệ thực vật - CĐ) - Trường Cao đẳng Đà Lạt [Mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260224/hoacattuong2026/135x160/32161772096417.jpg)
