
Structural insights into mechanisms of non-nucleoside
drug resistance for HIV-1 reverse transcriptases mutated
at codons 101 or 138
Jingshan Ren
1
, Charles E. Nichols
1
, Anna Stamp
1
, Phillip P. Chamberlain
1
, Robert Ferris
2
,
Kurt L. Weaver
2
, Steven A. Short
2
and David K. Stammers
1
1 Division of Structural Biology, The Wellcome Trust Centre for Human Genetics, Henry Wellcome Building for Genomic Medicine,
University of Oxford, UK
2 Glaxo Smith Kline Inc., Research Triangle Park, NC, USA
The emergence of resistant viruses resulting from drug
treatment of HIV-infected patients poses one of the
most significant problems in countering AIDS in West-
ern countries [1]. Two virus specific proteins, reverse
transcriptase (RT) and protease, have been the main
targets for the development of anti-HIV drugs used
in multidrug combination therapy regimens [2]. HIV-1
RT contains two distinct inhibitor binding sites for
Keywords
drug resistance; HIV-1 reverse transcriptase
mutants; Lys101Glu, Glu138Lys; non-
nucleoside inhibitors; X-ray crystallography
Correspondence
D.K. Stammers, Division of Structural
Biology, The Wellcome Trust Centre for
Human Genetics, University of Oxford,
Roosevelt Drive, Oxford OX3 7BN, UK
Fax: +44 1865 287 547
Tel: +44 1865 287 565
E-mail: daves@strubi.ox.ac.uk
(Received 23 March 2006, revised 20 June
2006, accepted 22 June 2006)
doi:10.1111/j.1742-4658.2006.05392.x
Lys101Glu is a drug resistance mutation in reverse transcriptase clinically
observed in HIV-1 from infected patients treated with the non-nucleoside
inhibitor (NNRTI) drugs nevirapine and efavirenz. In contrast to many
NNRTI resistance mutations, Lys101(p66 subunit) is positioned at the sur-
face of the NNRTI pocket where it interacts across the reverse transcrip-
tase (RT) subunit interface with Glu138(p51 subunit). However, nevirapine
contacts Lys101 and Glu138 only indirectly, via water molecules, thus the
structural basis of drug resistance induced by Lys101Glu is unclear. We
have determined crystal structures of RT(Glu138Lys) and RT(Lys101Glu)
in complexes with nevirapine to 2.5 A
˚, allowing the determination of water
structure within the NNRTI-binding pocket, essential for an understanding
of nevirapine binding. Both RT(Glu138Lys) and RT(Lys101Glu) have
remarkably similar protein conformations to wild-type RT, except for sig-
nificant movement of the mutated side-chains away from the NNRTI
pocket induced by charge inversion. There are also small shifts in the posi-
tion of nevirapine for both mutant structures which may influence ring
stacking interactions with Tyr181. However, the reduction in hydrogen
bonds in the drug-water-side-chain network resulting from the mutated
side-chain movement appears to be the most significant contribution to
nevirapine resistance for RT(Lys101Glu). The movement of Glu101 away
from the NNRTI pocket can also explain the resistance of RT(Lys101Glu)
to efavirenz but in this case is due to a loss of side-chain contacts with the
drug. RT(Lys101Glu) is thus a distinctive NNRTI resistance mutant in
that it can give rise to both direct and indirect mechanisms of drug resist-
ance, which are inhibitor-dependent.
Abbreviations
NRTI, nucleoside analogue inhibitors of RT; NNRTI, non-nucleoside reverse transcriptase inhibitor; NtRTI, nucleotide analogue; PETT,
phenethylthiazolylthiourea; RT, reverse transcriptase; TSAO, (2¢,5¢-bis-O-(tert-butyldimethylsilyl)-b-d-ribofuranosyl]-3¢-spiro-5¢¢-(4¢¢-amino-
1¢¢,2¢¢-oxathiole-2¢¢,2¢¢-dioxide).
3850 FEBS Journal 273 (2006) 3850–3860 ª2006 The Authors Journal compilation ª2006 FEBS

different classes of drugs within the larger subunit of
the p66 ⁄p51 heterodimer [3,4]. Nucleoside analogue
inhibitors of RT (NRTIs), such as zidovudine, lamivu-
dine, stavudine and the nucleotide analogue (NtRTI)
tenofovir, bind, respectively, as activated tri- or di-
phosphate forms at the RT polymerase active site.
NRTIs and NtRTIs, as well as competing with cognate
nucleotide substrates, are also incorporated into the
primer strand causing termination of the growing
DNA chain [5]. Non-nucleoside inhibitors (NNRTIs)
form a second class of compounds that target HIV-1
RT [6]. NNRTIs bind at an allosteric site about 10 A
˚
from the polymerase active site. The structural mech-
anism of NNRTI inhibition involves distortion of the
catalytically essential triad of aspartic acid residues
[7,8]. The non-nucleoside site, although almost entirely
contained within the p66 subunit, also has Glu138
from the p51 subunit located at the edge of the inhib-
itor pocket [3,4]. First-generation NNRTI drugs such
as nevirapine (Scheme 1) and delavirdine generally
show significant reduction in potency in the presence
of a wide range of single point mutations [9]. Second
generation compounds, including efavirenz, have
greater resilience to many such mutations [10,11].
Whilst the majority of the mutation sites encoding
resistance to NRTIs are distal to the dNTP site within
RT [12,13], those which result in NNRTI drug resist-
ance are generally proximal to the inhibitor binding
pocket [3,4].
Many of the most commonly observed NNRTI
resistance mutations are located at hydrophobic resi-
dues at the centre of the drug binding pocket (e.g.
Leu100, Val106, Val108, Tyr181 and Tyr188). How-
ever, charged residues positioned towards the surface
of the drug pocket are also known sites of drug
resistance mutations. Indeed, Lys103Asn is the muta-
tion most frequently observed in clinical use of
NNRTIs. Other NNRTI resistance mutations at sur-
face residues in RT include Lys101 and Glu138 (the
latter residue from the p51 subunit). Mutations at
residues 101 and 138 that give resistance to NNRTIs
usually involve a change in the sign of the side-chain
charge, e.g. Lys101Glu or Glu138Lys ⁄Arg. Pyridinone
NNRTIs can select for the Lys101Glu mutation in
HIV-1 RT, giving approximately eight-fold resistance
to compounds in this series [14]. The Lys101Glu
mutation in RT also gives eight-fold resistance to
nevirapine, based on the mean of four published
values [15–18]. Significantly, Lys101Glu in RT is
observed in the clinic for drug regimens which include
nevirapine [19,20] or efavirenz [21] as the NNRTI
component. The Lys101Glu mutation in RT is also
selected by passaging HIV in tissue culture in the
presence of carboxanilide NNRTIs such as UC-38
[22], giving greater than 100-fold resistance [23]. In
the crystal structure with wild-type HIV-1 RT, UC-38
has a van der Waals contact with the side-chain of
Lys101 [24]. Consistent with the reports of the selec-
tion of Lys101Glu in clinical use of efavirenz, the
crystal structure of RT with this NNRTI shows a
contact of the inhibitor with the CD atom of Lys101
[25]. In the case of nevirapine, however, there are
only indirect contacts of Lys101 in the complex with
wild-type RT, via a series of three water molecules
that link the drug, the side-chain of Glu138 and the
main-chain carbonyl of Lys101 [4]. The molecular
mechanism of drug resistance for nevirapine induced
by Lys101Glu mutant is thus unclear, although an
indirect mechanism is present.
The RT(Glu138Lys) mutation in HIV is selected
in tissue culture by inhibitors of the TSAO (2¢,5¢-bis-
O-(tert-butyldimethylsilyl)-b-d-ribofuranosyl]-3¢-spiro-
5¢¢-(4¢¢-amino-1¢¢,2¢¢-oxathiole-2¢¢,2¢¢-dioxide) series [26].
TSAO compounds have been shown to be capable of
disrupting the p66 ⁄p51 heterodimer [27], indeed model-
ling studies suggest that certain TSAO NNRTIs may
bind at a novel site between the subunits of the RT
heterodimer [28]. It is proposed that mutation of
Glu138Lys causes loss of a key interaction of the gluta-
mic acid carboxyl group to an amino group of TSAO
inhibitors. Mutations in RT at codon 138 (to Lys ⁄Arg)
have been shown to give cross resistance to a number
of NNRTIs including members of the PETT series [29]
as well as to emivirine [17]. In contrast, RT(Glu138Lys)
is reported as having minimal cross resistance to nevi-
rapine [18,30]. PETT-2 (Scheme 1) forms a van der
Waals contact with the carboxyl group of Glu138 in
the complex with wild-type RT [31].
To date, crystal structure determinations of
NNRTI resistant HIV-1 RTs have mainly focussed
on mutations at positions 103, 181 and 188 [25,32–
36]. More recently structures of RT with drug resist-
ance mutations Leu100Ile, Val106Ala and Val108Ile
have been reported, which indicate an indirect com-
ponent in the mechanism of resistance via modulation
NN
O
N
N
Nevirapine
N
N
H
N
H
N
Cl
O
SF
PETT-2
H
1
10
Scheme 1. Structures of nevirapine and PETT-2 are shown.
J. Ren et al. HIV-1 RT drug resistance mutations at 101 and 138
FEBS Journal 273 (2006) 3850–3860 ª2006 The Authors Journal compilation ª2006 FEBS 3851

of interactions with the key aromatic side-chains of
Tyr181 and Tyr188 [37]. For RT(Lys103Asn) it has
been shown that the second generation NNRTI efavi-
renz can bind to the enzyme in both a wild-type con-
formation as well as with Tyr181 in a rearranged
position [25,36]. The side-chain of Asn103 can form a
hydrogen bond to the Tyr188 hydroxyl thereby stabil-
izing the unliganded RT structure [7,35]. In work
reported here, crystal structures of HIV-1 RTs either
containing the mutation Lys101Glu or Glu138Lys in
complexes with nevirapine and PETT-2 have been
determined in order to investigate structural effects
of such mutants on the drug binding pocket and
how these might relate to resistance mechanisms.
RT(Glu138Lys) and RT(Lys101Glu) mutants are logi-
cal to study as a pair as they are both charged sur-
face residues interacting across the p66 ⁄p51 interface.
The availability of a range of structures of NNRTI-
resistant RTs drugs will provide greater understanding
of the molecular basis of drug resistance mechanisms.
Such data will contribute to the design of novel inhibi-
tors, which are still needed for containing the effects of
HIV infection.
Results
Overall RT structure
Details of the X-ray data collection and the
refinement statistics are shown in Table 1. Of the
wide range of NNRTIs tested for co-crystallization
with RT(Glu138Lys) and RT(Lys101Glu), three
complexes gave crystals suitable for structure
determination: RT(Lys101Glu)-nevirapine (to 2.5-A
˚
resolution), RT(Glu138Lys)-nevirapine (2.5 A
˚) and
RT(Glu138Lys)-PETT2 (3.0 A
˚). The relatively high
resolution of the mutant RT-nevirapine structures
allowed details of the water structure associated with
the protein relevant to NNRTI binding to be deter-
mined. Water molecules are important for the interac-
tions of nevirapine with RT, particularly via Lys101
and Glu138, as there are no direct contacts of the
inhibitor with these side-chains. Fo-Fc omit maps
show clear electron density for the bound NNRTI,
some water molecules, as well as for the mutated side-
chains (Fig. 1) in each case. Side-chain electron density
relevant to NNRTI drug resistance is also shown in
Table 1. Statistics for crystallographic structure determinations.
Data collection details
Data set K101E-nevirapine E138K-nevirapine E138K-PETT-2
Data collection site PF BL-6 A APS SBC-2 SRS PX14.2
Detector Fuji BAS III CCD SBC CCD ADSC-Q4
Wavelength (A
˚) 1.000 0.82657 0.979
Unit cell dimensions (a,b,c in A
˚)
a
138.9, 114.9, 65.6 139.6, 115.0, 65.6 138.8, 114.8, 65.9
Resolution range (A
˚) 30.0–2.5 30.0–2.5 30.0–3.0
Observations 149821 251696 57571
Unique reflections 35057 37104 19095
Completeness (%) 94.4 99.5 88.0
Average I⁄r(I) 9.8 12.6 11.8
R
mergeb
0.100 0.089 0.083
Outer resolution shell
Resolution range (A
˚) 2.59–2.50 2.59–2.50 3.11–3.00
Unique reflections 3371 3627 1537
Completeness (%) 92.5 99.5 72.6
Average I⁄r(I) 1.0 1.1 1.4
Refinement statistics:
Resolution range (A
˚) 30.0–2.5 30.0–2.5 20.0–3.0
Number of reflections(working ⁄test) 33264 ⁄1740 35199 ⁄1856 18153 ⁄929
R-factor
c
(R
work
⁄R
free
) 0.198 ⁄0.270 0.216 ⁄0.288 0.226 ⁄0.277
R-factor
c
(all data) 0.194 0.203 0.226
Number of atoms (protein ⁄inhibitor ⁄water) 7570 ⁄20 ⁄80 7614 ⁄20 ⁄79 7554 ⁄23 ⁄–
Rms bond length deviation (A
˚) 0.008 0.008 0.009
Rms bond angle deviation () 1.4 1.4 1.5
Mean B-factor (A
˚
2
)
d
59 ⁄65 ⁄37 ⁄44 62 ⁄68 ⁄39 ⁄48 73 ⁄81 ⁄60 ⁄–
Rms backbone B-factor deviation (A
˚
2
) 4.4 4.2 3.4
a
All crystals belong to space group P2
1
2
1
2
1
[40,41];
b
R
merge
¼S|I–<I>|⁄S <I>;
c
R factor ¼S|F
o
-F
c
|⁄SF
o
;
d
mean B factor for main-chain,
side-chain, inhibitor and water molecules.
HIV-1 RT drug resistance mutations at 101 and 138 J. Ren et al.
3852 FEBS Journal 273 (2006) 3850–3860 ª2006 The Authors Journal compilation ª2006 FEBS
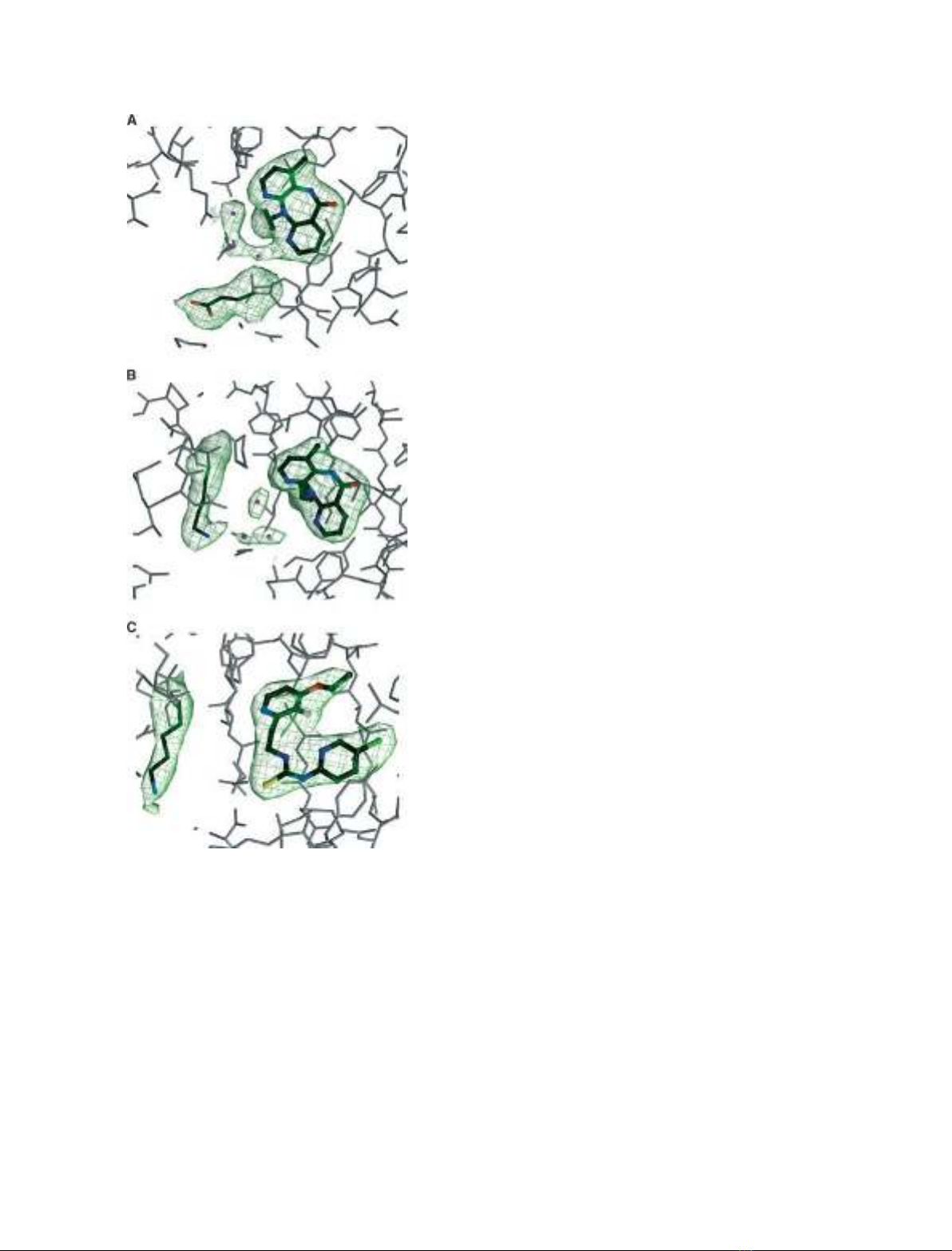
Fig. 1, i.e. for Lys101Glu in the p66 subunit (Fig. 1A)
and Glu138Lys in the p51 subunit (Fig. 1B,C).
Comparison of wild-type and mutant RT structures
Lys101Glu
In the crystal structure of RT(Lys101Glu) in complex
with nevirapine, electron density for the mutant gluta-
mic acid side-chain is well defined (Fig. 1A). The struc-
ture of the NNRTI pocket is overall remarkably
similar for mutant and wild-type, with the exception of
the mutated side-chain and adjacent interacting resi-
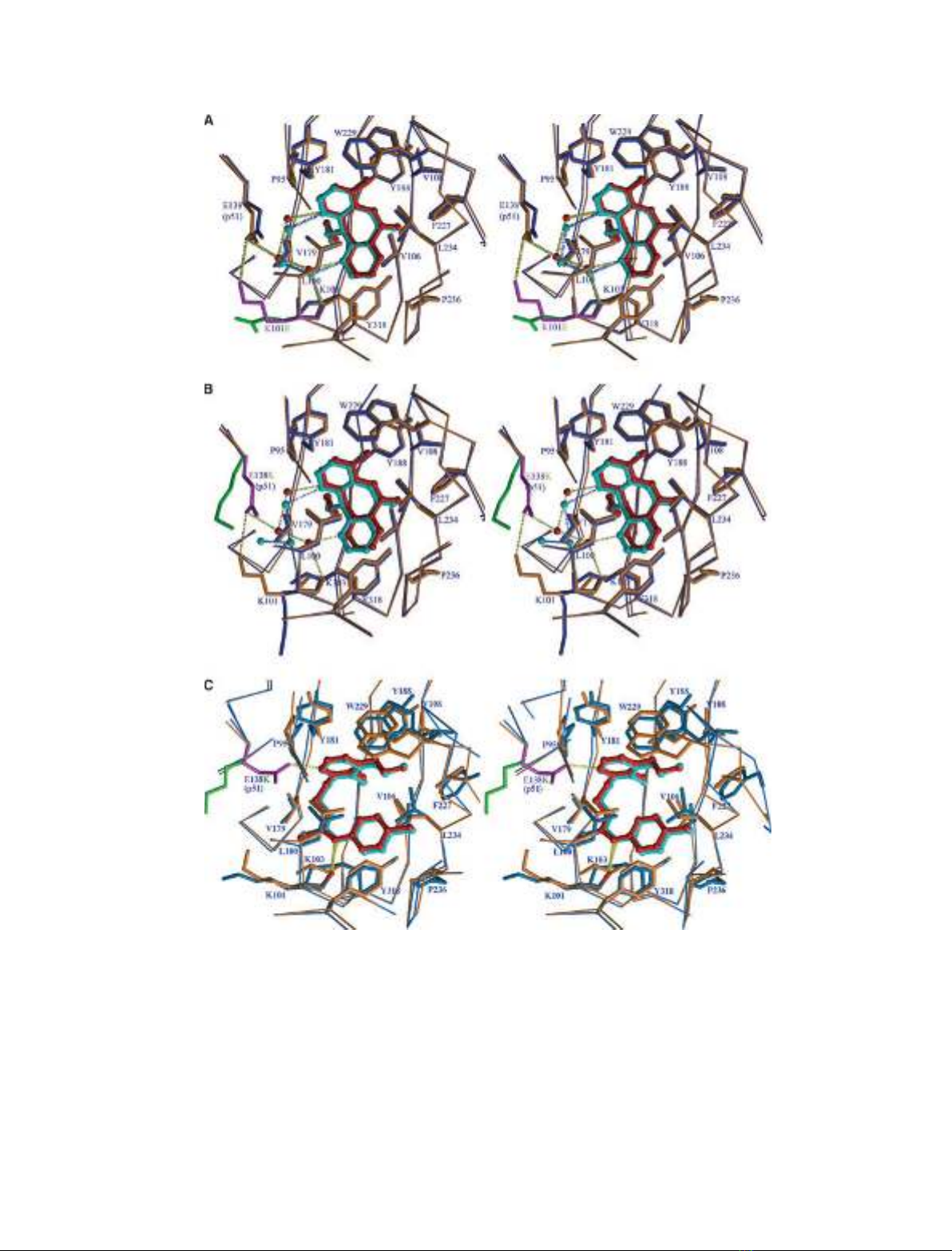
due. There is a significant movement of Glu101 relat-
ive to the position of the wild-type Lys101, such that
the side-chain carboxyl group points away from the
NNRTI site (distance of the carboxyl group to the
equivalent wild-type lysine side-chain amino group of
3.4 A
˚) (Fig. 2A). There is also a smaller displacement
of the Glu138 side-chain with a shift of 0.5 A
˚relative
to wild-type. The result of these changes is the loss of
the salt bridge that links the protonated amino group
of Lys101 in the p66 subunit to the negatively charged
carboxyl of the Glu138 side-chain from the p51 sub-
unit in the wild-type RT nevirapine complex [4].
Accompanying the side-chain movement related to the
Lys101Glu mutation there is also a shift in the posi-
tion of nevirapine, with an average atom displacement
of 0.3 A
˚and a maximum movement of 0.5 A
˚. The
side-chains of Tyr181 and Tyr188 are however, main-
tained in a wild-type conformation. In spite of the
observed rearrangements of inhibitor and the side-
chains of residues 101 and 138, the three water mole-
cules that link nevirapine to Glu138 in the wild-type
complex are retained in the mutant structure albeit
with shifts in position in the same direction as the
nevirapine, i.e. away from the binding pocket.
Glu138Lys
The structures of HIV-1 RT(Glu138Lys) in complexes
with nevirapine and PETT-2 complex have well defined
side-chain density for Lys138 (p51) (Fig. 1B,C). As
with the Lys101Glu mutant, the overall structure of
the NNRTI pocket in RT(Glu138Lys) closely matches
that of the equivalent nevirapine complex with wild-
type RT, with the exception of the mutated side-chain
itself, which together with Lys101 undergoes large
positional changes. Lys138 moves away from the
NNRTI pocket in the complex with nevirapine as indi-
cated by the shift in position of the CD atom of 2.8 A
˚
(Fig. 2B), whilst Lys101 becomes much less ordered
than in the wild-type structure. The best fit of the side-
chain to electron density indicates a rotation of close
to 150about the Lys101 CA–CB bond, positioning it
away from the NNRTI pocket (Fig. 2B) and resulting
in the loss of interaction of residue 138 in p51 with
Lys101 in the p66 subunit. There is also a shift in the
position of nevirapine outwards from the binding
pocket with an average atom displacement of 0.4 A
˚
and a maximum displacement of 0.6 A
˚. The three
water molecules that bridge between Glu138 and
Fig. 1. Simulated annealing omit electron density maps contoured
at 3.5rshowing bound inhibitors, waters and mutated residues
within one of the HIV-1 RT subunits as indicated. (A) Lys101Glu in
p66 and nevirapine. (B) Glu138Lys in p51 and nevirapine. (C)
Glu138Lys in p51 and PETT-2.
J. Ren et al. HIV-1 RT drug resistance mutations at 101 and 138
FEBS Journal 273 (2006) 3850–3860 ª2006 The Authors Journal compilation ª2006 FEBS 3853
nevirapine in the wild-type complex remain in the
Glu138Lys mutant but are displaced in position and
the network of hydrogen bonds is altered. The remain-
ing side-chains within the mutant NNRTI binding site
are essentially positioned the same as for the wild-type
enzyme.
Fig. 2. Stereo-diagrams comparing the NNRTI binding sites of wild-type and mutant RTs for the following complexes: (A) Lys101Glu and
nevirapine, (B) Glu138Lys and nevirapine, and (C) Glu138Lys and PETT-2. The thinner and thicker bonds show the CA backbone and side-
chains with wild-type RT coloured orange and the mutant RTs coloured blue, respectively. Inhibitors and water molecules are shown in ball-
and-stick representation and coloured red for wild-type RT and cyan for mutant RTs. For clarity, the side-chains at the site of mutation are
shown in magenta for wild-type and green for the mutants. Hydrogen bonds linking drug to protein and drug to water molecules are marked
in dashed lines, coloured yellow for wild-type and blue for mutant RTs.
HIV-1 RT drug resistance mutations at 101 and 138 J. Ren et al.
3854 FEBS Journal 273 (2006) 3850–3860 ª2006 The Authors Journal compilation ª2006 FEBS


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)