
257
CÁC ĐẶC TRƯNG CẤU TRÚC VÀ ĐỘNG HỌC
CỦA VẬT LIỆU ĐA TINH THỂ BẠC (Ag)
Nguyễn Thị Huỳnh Nga 1, Mai Văn Dũng1
1. Trường Đại học Thủ Dầu Một.
TÓM TẮT
Trong nghiên cứu này, chúng tôi sử dụng phương pháp mô phỏng động lực học phân tử
đ khảo sát cấu trúc của vật liệu đa tinh th Ag dưới ảnh hưởng của nhiệt độ. Các đặc trưng
cấu trúc của hệ đa tinh th Ag bao gồm 16383 nguyên tử với thế tương tác EAM được phân tích
thông qua tổng năng lượng trên mỗi nguyên tử, nhiệt dung riêng, hàm phân bố xuyên tâm, sự
thay đổi của các đơn vị cấu trúc và phân bố góc. Các kết quả mô phỏng cho thấy nhiệt độ nóng
chảy của cấu trúc đa tinh th Ag là 1500
50 K. Động học của hệ cũng được thảo luận trong
nghiên cứu này thông qua độ dịch chuyn bình phương trung bình và hệ số khuếch tán của các
nguyên tử Ag. Các kết quả nghiên cứu cung cấp những thông tin cần thiết cho các nghiên cứu
thực nghiệm.
Từ khoá: cấu trúc; động lực học; đặc trưng; đa tinh th, mô phỏng.
1. TỔNG QUAN
Bạc (Ag) là kim loại chuyển tiếp quý có màu trắng, bóng, mềm, nhẹ, dễ uốn, có tính dẫn
điện và nhiệt cao. Ag đã được biết đến rộng rãi do có nhiều lợi ích và gần gũi với con người.
Người ta sử dụng Ag dưới nhiều hình thức như đồng xu, bình, giấy bạc, chỉ khâu và chất keo
như kem dưỡng da, thuốc bôi, v.v. Các đặc tính chữa bệnh của Ag cũng được nghiên cứu trong
hơn 2000 năm qua. Từ thế kỷ 19, các hợp chất gốc bạc đã được sử dụng trong ứng dụng kháng
khuẩn (Hayelom Dargo Beyene và nnk, 2017; Edwards & Petersen, 1936). Bạc được sử dụng
trong việc chữa lành vết thương (G. Nam và nnk, 2016), nhiều ứng dụng y sinh (Ekaterina O.
Mikhailova, 2020). Ngoài ra, Ag còn được ứng dụng nhiều trong khoa học và công nghệ (Bryan
Calderón-Jiménez và nnk, 2017) như: vật liệu siêu dẫn (Hui Jiang và nnk, 2020), vật liệu kháng
khuẩn (Vukoman Jokanović và nnk, 2024), cảm biến và vi điện tử (ZD Lin và nnk, 2013), chất
xúc tác quang (D. M. Tobaldi, C. Piccirillo, 2014) và vật liệu từ tính (George V. Belessiotis ,
Pinelopi P. Falara, 2022; 15. Asmaa. A. H. El-Bassuony và nnk, 2023). Các phương pháp để
chế tạo và nghiên cứu tính chất của Ag như phương pháp lý thuyết, phương pháp thực nghiệm
hoặc phương pháp mô phỏng. Đối với các nghiên cứu thực nghiệm, hạt nano Ag đã được tổng
hợp bằng phương pháp hóa học và phương pháp sinh học (Krishna Gudikandula & Singara
Charya Maringanti, 2016). Đối với các nghiên cứu lý thuyết, cụ thể như phương pháp lượng tử
hóa có thể được sử dụng để nghiên cứu Ag (Bryan Calderón-Jiménez, et al, 2017), nhưng
phương pháp mô phỏng được coi là phương pháp ưu việt nhất, với khả năng mô phỏng vật liệu
Ag ở cấp độ nguyên tử. Phương pháp động lực phân tử (MD) được sử dụng để nghiên cứu các
đặc tính cấu trúc, nhiệt động của vật liệu (Akbarzadeh và nnk, 2014; Hamed Akbarzadeh,
Hamzeh Yaghoubi, 2013; Plimpton, S, 1995; R.A. Johnson, 1989). Kết quả cho thấy nhiệt độ
chuyển tiếp (Tm) giảm khi kích thước của vật liệu giảm (Asoro MA và nnk, 2010; Wenhua Luo,
et al, 2008), sự xuất hiện của cấu trúc thập diện và cấu trúc nhị thập diện rất thú vị. Ngoài ra,
mối quan hệ giữa kích thước và nhiệt độ Kauzmann (TK), cũng như mối quan hệ giữa nhiệt độ

258
nóng chảy (Tm), entanpy nóng chảy (Hm) và entropy nóng chảy (Sm) (Shifang Xiao và nnk,
2005; H. A. Alarifi, et al, 2013; Y. F. Zhu và nnk, 2009), có thể quan sát trực quan mô hình cấu
trúc bằng phần mềm OVITO (Stukowski A, 2009). Tương tự, Kuzmin và cộng sự nghiên cứu
vật liệu Ag trong khoảng nhiệt độ từ 0K đến 1300K bằng phương pháp động lực phân tử (MD)
sự chuyển đổi cấu trúc bát diện sang cấu trúc đa diện (V. I. Kuzmin và nnk, 2008). Nghiên cứu
của Tian và cộng sự cho thấy số lượng nguyên tử khác nhau dẫn đến đặc điểm cấu trúc khác
nhau (Tian và nnk, 2009; S. Jalili và nnk, 2012). Kết quả cũng cho thấy hạt nano Ag có xu
hướng kết tinh ở nhiệt độ (Tg), Tg = 453K (Hongjun Ji và nnk, 2014) và quá trình chuyển pha
xảy ra ở nhiệt độ chuyển pha (Tm), Tm = 1234K (Dung T. N và nnk, 2020; Qingshan Fu và nnk,
2016; Lumsdon and Caroline, 2021; M. P. Samantaray and S.S. Sarangi, 2020). Việc giảm kích
thước của các hạt dẫn đến giảm entanpy và entropy của sự nóng chảy, dẫn đến hiệu ứng tăng
kích thước (Qingshan Fu và nnk, 2016). Nghiên cứu lý thuyết cũng cho thấy năng lượng kết
tinh phụ thuộc vào kích thước, nhiệt độ nóng chảy và nhiệt độ Debye (Y. F. Zhu và nnk, 2009;
Chakravarty and Charusita, 2007). Từ những vấn đề nói trên, một câu hỏi đặt ra là các yếu tố
ảnh hưởng đến sự thay đổi cấu trúc và đặc biệt là cấu trúc của vật liệu đa tinh thể. Bài viết này
chúng tôi sử dụng phương pháp mô phỏng động lực học phân tử để khảo sát cấu trúc của vật
liệu đa tinh thể Ag. Mô hình phân tử cỡ 1600 nguyên tử với thế tương tác EAM. Kết quả mô
phỏng sẽ cung cấp những thông tin cần thiết về tổng năng lượng trên mỗi nguyên tử, nhiệt dung
riêng, hàm phân bố xuyên tâm, phân bố góc, độ dịch chuyển bình phương trung bình, hệ số
khuếch tán, sự thay đổi các đơn vị cấu trúc cho các nghiên cứu thực nghiệm.
2. PHƯƠNG PHÁP TÍNH TOÁN
2.1. Xây dựng mô hình tính toán
Cấu trúc đa tinh thể Ag bao gồm 16383 nguyên tử với kích thước tương ứng là 65.368
65.368 65.368 Å3. Mô hình cấu trúc như hình 2.1, trong đó các nguyên tử FCC và Other Ag
lần lượt là màu xanh và màu đỏ.
Hình 2.1. Mô hình cấu trúc đa tinh th của vật liệu Ag.
2.2. Phương pháp mô phỏng
Trong bài báo này, sự thay đổi cấu trúc của hệ đa tinh thể Ag được khảo sát bằng việc sử
dụng phương pháp mô phỏng động lực học phân tử với điều kiện biên tuần hoàn được áp dụng
cho cả ba hướng x, y, z. Chúng tôi sử dụng phần mềm LAMMPS để khảo sát các đặc trưng cấu
trúc của hệ đa tinh thể Ag (Hamed Akbarzadeh and Hamzeh Yaghoubi, 2013). Để giải gần đúng
phương trình chuyển động của các nguyên tử, chúng tôi sử dụng thuật toán Verlet với bước thời
259
gian mô phỏng là 1.0 fs. Thế tương tác EAM đã được Johnson sử dụng rất thành công khi thực
hiện khảo sát các tính chất của các hệ hợp kim có cấu trúc FCC, biểu thức năng lượng của thế
EAM được mô tả như biểu thức (2.1) (R.A. Johnson, 1989).
𝐸 = ∑{𝐹𝑖(𝜌𝑖
)+1
2∑𝑉
𝑖𝑗(𝑟𝑖𝑗)
𝑖≠𝑗 }
𝑖 (2.1)
trong đó F là hàm của mật độ trung bình electron 𝜌𝑖
, V𝑖𝑗(𝑟𝑖𝑗) là tương tác cặp giữa các nguyên
tử i và j ở khoảng cách 𝑟𝑖𝑗. Để tính toán phân bố số phối trí, chúng tôi chọn bán kính cắt là 3.95
Å, đây là vị trí thấp nhất sau đỉnh đầu tiên của hàm phân bố xuyên tâm ở nhiệt độ 3000K. Để
khảo sát sự thay đổi cấu trúc của vật liệu đa tính thể Ag, chúng tôi thực hiện như sau: Ban đầu
mô hình đa tinh thể Agđược tạo ra ở 300 K bằng việc hồi phục trong khoảng thời gian 100ps
cho đến khi nó đạt trạng thái cân bằng ở áp suất 0 GPa. Sau đó mô hình được nung nóng đến
3000K với tốc độ 1012 K/s. Các đặc trưng cấu trúc, độ dịch chuyển bình phương trung bình và
trực quan mô hình được hồi phục trong khoảng thời gian 100ps. Phần mềm OVITO được sử
dụng để trực quan mô hình và tính toán các đôn vị cấu trúc bằng phương pháp Common
Neighbor Analysis (CNA) (Stukowski A, 2009).
3. KẾT QUẢ VÀ THẢO LUẬN
3.1. Các đặc trưng cấu trúc của vật liệu khi nhiệt độ thay đổi
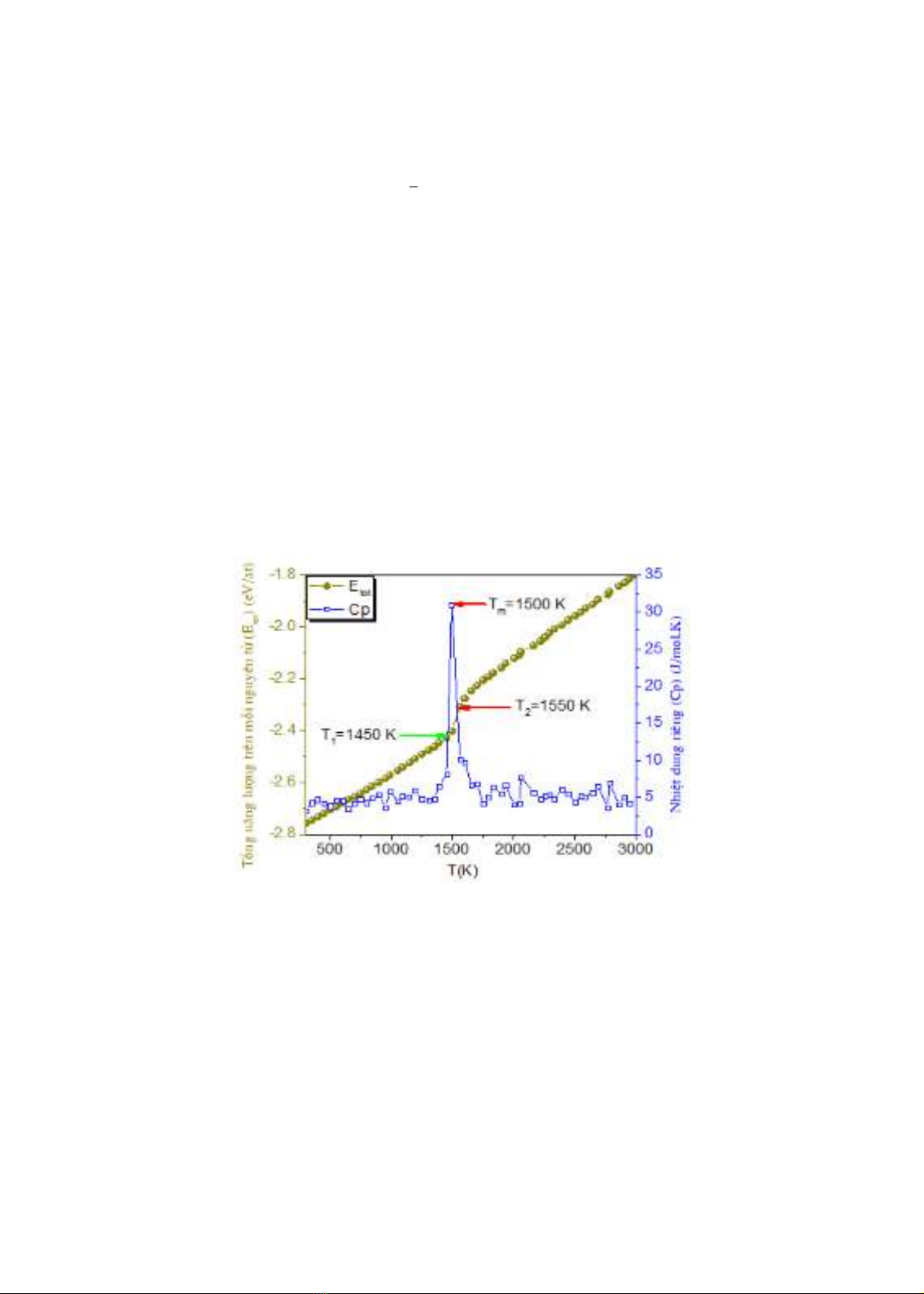
Hình 3.1. Sự thay đổi của tổng năng lượng trên mỗi nguyên tử theo nhiệt độ.
Hình 3.1 mô tả sự thay đổi của thế năng trên mỗi nguyên tử khi nhiệt độ tăng từ 300K
đến 3000K. Chúng ta có thể thấy rằng trong khoảng nhiệt độ từ 300K đến 1450K, thế năng trên
mỗi nguyên tử biến đổi tuyến tính với nhiệt độ, ở trong vùng nhiệt độ này các nguyên tử chủ
yếu thực hiện dao động nhiệt xung quanh vị trí cân bằng của chúng. Điều này cho thấy rằng mô
hình đang ở trạng thái rắn tinh thể. Ở nhiệt độ 1450K, thế năng trên mỗi nguyên tử tăng đột
ngột và biến đổi tuyến tính với nhiệt độ cho đến nhiệt độ 1550K. Điều này có nghĩa là mô hình
đã chuyển sang trạng thái nóng chảy. Như vậy quá trình nóng chảy diễn ra trong khoảng nhiệt
độ từ 1450K đến 1550K. Ở vùng nhiệt độ lớn hơn 1550K, thế năng lại tăng vọt và biến đổi
tuyến tính với nhiệt độ đến 3000K. Điều này chỉ ra rằng mô hình đã chuyển sang trạng thái
nóng chảy hoàn toàn. Như vậy, nhiệt độ nóng chảy của mô hình được xác định là Tm= (1450
+1550 )/2=1500K. Giá trị này cao hơn so với kết quả thực nghiệm của nhóm tác giả (M. P.
Samantaray and S.S. Sarangi, 2020) và kết quả mô phỏng gần với kết quả mô phỏng của Dung
260
và cộng sự (Dung T. N và nnk, 2020). Cần chú rằng các kết quả tính toán trong nghiên cứu của
chúng tôi được thực hiện trong không gian hai chiều và bề mặt tự do. Ngoài ra, nhiệt độ nóng
chảy của mô hình cũng được xác định thông qua việc tính toán nhiệt dung riêng được mô tả
như hình 3.1 (đường màu xanh). Nhiệt dung riêng có thể được tính toán thông qua hệ thức
Cp=∆E/∆T, trong đó ∆T bằng 50 K. Có thể thấy rằng nhiệt dung riêng có một đỉnh cao nhất ở
vị trí có nhiệt độ Tm=1500 K. Như vậy khi nóng chảy nhiệt dung riêng tăng vọt, cho thấy đây
là chuyển pha loại một. Ở vùng nhiệt độ lớn hơn Tm và nhỏ hơn Tm, nhiệt dung riêng Cp có độ
cao các đỉnh rất nhỏ và dao động nhẹ. Điều này có nghĩa Tm là nhiệt độ nóng chảy của hệ.
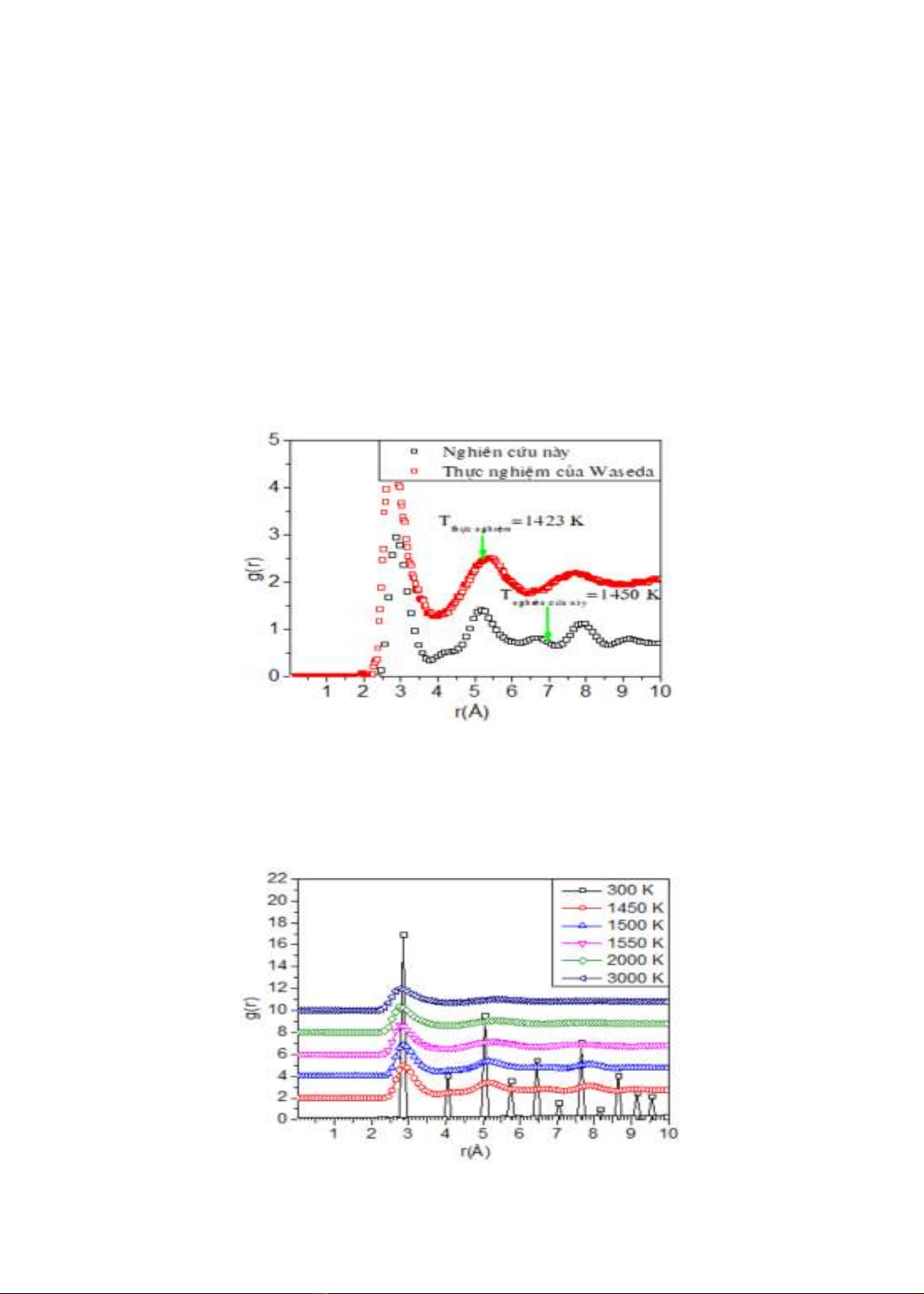
Để đánh giá độ tin cậy của mô hình, chúng tôi thực hiện so sánh hàm phân bố xuyên tâm
ở nhiệt độ 1450K với kết quả thực nghiệm của Waseda ở nhiệt độ 1423 K như hình 3.2 (Y.
Waseda and M. Ohtani, 1974). Kết quả cho thấy vị trí đỉnh đầu tiên phù hợp với kết quả thực
nghiệm của Waseda, tuy nhiên độ cao của đỉnh thấp hơn so với kết quả thực nghiệm. Điều này
có thể liên quan đến thế tương tác được sử dụng trong mô phỏng của chúng tôi, cũng như các
điều kiện thực hiện thí nghiệm.
Hình 3.2. So sánh hàm phân bố xuyên tâm của nghiên cứu này với thực nghiệm.
Để tiếp tục khảo sát sự thay đổi cấu trúc của hạt tinh thể Ag, chúng tôi phân tích hàm
phân bố xuyên tâm (PBXT) ở các nhiệt độ khác nhau. Trước tiên, chúng tôi thực hiện so sánh
hàm phân bố xuyên tâm ở nhiệt độ nóng chảy với kết quả thực nghiệm của Dung (Dung. T. N
và nnk, 2020).
Hình 3.3. Hàm phân bố xuyên tâm ở các nhiệt độ khác nhau.
261
Sự thay đổi cấu trúc của vật liệu dưới ảnh hưởng của nhiệt độ cũng được xác định thông
qua hàm PBXT như trên hình 3.3. Nhìn chung khi nhiệt độ tăng PBXT của các cặp nguyên tử
đều có độ cao đỉnh giảm xuống. Có thể thấy rằng ở nhiệt độ thấp 300K, hàm PBXT của các cặp
Ag-Ag có các đỉnh nhọn và có độ cao rất lớn. Điều này cho thấy mô hình đang ở trạng thái rắn.
Khi nhiệt độ tăng đến 1450K, độ cao của các đỉnh giảm và mở rộng hơn, điều này cho thấy mô
hình bắt đầu chuyển sang trạng thái nóng chảy. Trong đó, vị trí của đỉnh thứ nhất của cặp Ag-
Ag là 2.89 Å. Sự nung nóng tiếp tục được tăng cường, độ cao đỉnh của PBXT giảm mạnh, đỉnh
thứ hai của cặp Ag-Ag là 5.1 Å và các đỉnh ở xa hơn gần như không còn tồn tại. Ở nhiệt độ
3000 K, hàm PBXT chỉ có một đỉnh duy nhất. Các kết quả này phù hợp với kết quả nghiên cứu
trong công trình của tác giả Dung và cộng sự. Điều này có nghĩa là mô hình đã chuyển sang
trạng thái nóng chảy hoàn toàn.
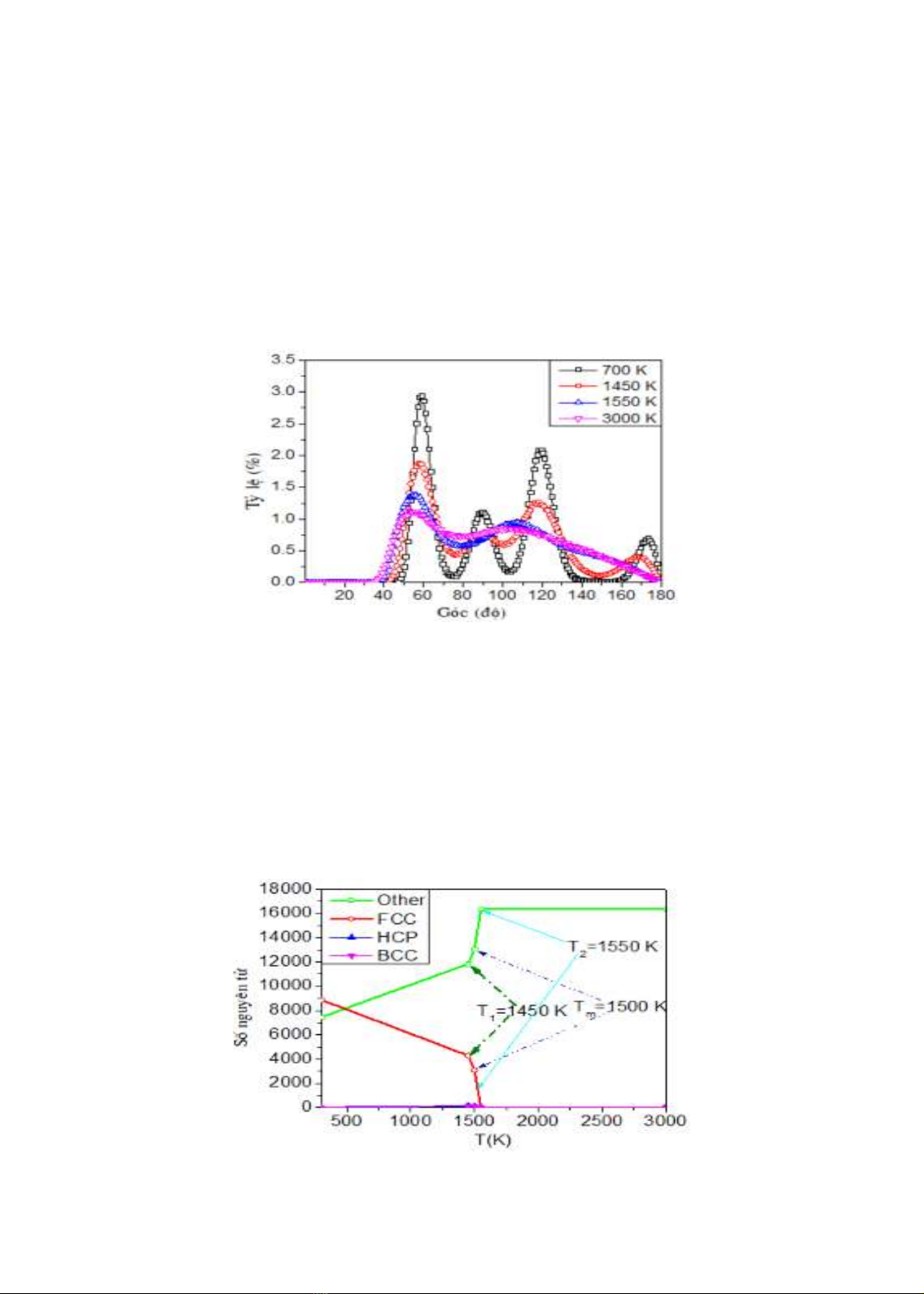
Hình 3.4. Phân bố góc xung quanh nhiệt độ nóng chảy.
Hình 3.4 là phân bố góc Ag-Ag-Ag xung quanh nhiệt độ nóng chảy của mô hình tinh thể
Ag. Có thể thấy rằng khi nhiệt độ tăng độ cao đỉnh phân bố góc Ag-Ag-Ag giảm mạnh. Từ hình
3.4 có thể thấy rằng, ở nhiệt độ 700K phân bố góc Ag-Ag-Ag có ba đỉnh nhọn và cao ở các vị
trí 60o, 90o và 120o. Điều này cho thấy rằng mô hình đang ở trạng thái rắn theo cấu trúc tinh thể
FCC ban đầu. Khi nhiệt độ tăng đến 1450K, độ cao của đỉnh phân bố góc Ag-Ag-Ag giảm
mạnh vùng phân bố mở rộng hơn rất nhiều và chỉ có hai đỉnh rõ ràng ở vị trí 60o và 120o có
nghĩa là mô hình bắt đầu chuyển sang trạng thái nóng chảy. Ở nhiệt độ 3000K độ cao đỉnh càng
mở rộng hơn và chỉ còn duy nhất một đỉnh rõ ràng ở vị trí 60o cho thấy mô hình đã chuyển sang
trạng thái nóng chảy hoàn toàn.
Hình 3.5. Sự thay đổi các đơn vị cấu trúc khi nhiệt độ thay đổi.

























