
Structure and topology of the transmembrane domain 4 of the
divalent metal transporter in membrane-mimetic environments
Hongyan Li
1,2
, Fei Li
1
, Zhong Ming Qian
2
and Hongzhe Sun
1
1
Department of Chemistry and Open Laboratory of Chemical Biology, The University of Hong Kong, China;
2
Department of
Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong, China
The divalent metal transporter (DMT1) is a 12-transmem-
brane domain protein responsible for dietary iron uptake in
the duodenum and iron acquisition from transferrin in
peripheral tissues. The transmembrane domain 4 (TM4)
of DMT1 has been shown to be crucial for its biological
function. Here we report the 3D structure and topology of
the DMT1-TM4 peptide by NMR spectroscopy with
simulated annealing calculations in membrane-mimetic
environments, e.g. 2,2,2-trifluoroethanol and SDS micelles.
The 3D structures of the peptide are similar in both envi-
ronments, with nonordered and flexible N- and C-termini
flanking an ordered helical region. The final set of the 16
lowest energy structures is particularly well defined in the
region of residues Leu9–Phe20 in 2,2,2-trifluoroethanol,
with a mean pairwise root mean square deviation of
0.23 ± 0.10 A
˚for the backbone heavy atoms and
0.82 ± 0.17 A
˚for all heavy atoms. In SDS micelles, the
length of the helix is dependent on pH values. In particular,
the C-terminus becomes well-structured at low pH (4.0),
whereas the N-terminal segment (Arg1–Gly7) is flexible and
poorly defined at all pH values studied. The effects of
12-doxylPtdCho spin-label and paramagnetic metal ions on
NMR signal intensities demonstrated that both the N-ter-
minus and helical region of the TM4 are embedded into the
interior of SDS micelles. Unexpectedly, we observed that
amide protons exchanged much faster in SDS than in 2,2,
2-trifluoroethanol, indicating that there is possible solvent
accessibility in the structure. The paramagnetic metal ions
broaden NMR signals from residues both situated in aque-
ous phase and in the helical region. From these results we
speculate that DMT1-TM4s may self-assemble to form a
channel through which metal ions are likely to be trans-
ported. These results might provide an insight into the
structure-function relationship for the integral DMT1.
Keywords:DMT1;membrane;NMR;structure.
The divalent metal transporter (DMT1) gene, also known as
Nramp2 (natural resistance-associated macrophage protein-
2) and DCT1, was identified recently [1,2]. It belongs to a
large family of integral membrane proteins highly conserved
throughout evolution, from bacteria to human beings [3–6].
It is the only known cellular iron importer, and is
responsible for importing iron from the gut into the entero-
cytes and also for transporting iron across the endosomal
membrane in the transferrin cycle [7–9]. The DMT1 consists
of 561 amino acids with 12 putative transmembrane
domains [1]. The DMT1 gene encodes two messenger
RNAs produced by alternative splicing of two 3¢exons that
show different 3¢untranslated regions containing an iron
response element (isoform I) and no iron response element
(isoform II), as well as distinct C-terminal protein sequences
[7–10]. Recently, DMT1 mRNA expression has also been
detected in the kidney [11].
Direct metal transport studies in Xenopus laevis oocytes
have demonstrated that DMT1 (isoform I) is a pH-
dependent divalent metal transporter with broad substrate
specificity including Fe
2+
,Mn
2+
,Co
2+
,Ni
2+
,Cu
2+
,Zn
2+
,
and toxic metals Cd
2+
and Pb
2+
[1]. Studies in cultured
mammalian cells have also shown that both isoforms of
DMT1 are capable of transporting a variety of divalent
metal ions across the plasma membrane [12,13]. Transport
of these metal ions was shown to occur at pH 5.5, but not at
7.4 [1]. The His267/His272 located in the transmembrane
domain (TM) 6 has been thought to play an important role
in pH regulation of metal transport by DMT1 [14].
However, it is not yet clear how pH regulates DMT1 metal
transport. The biological importance of this transporter is
shown by its involvement in two naturally occurring animal
mutants of iron metabolism. A mutation (G185R) in TM4
of DMT1 is responsible for microcytic anemia of the mk
mice and Belgrade rats, which exhibit severe defects in
intestinal iron absorption and erythroid iron utilization [2,7].
This suggests that the TM4 of DMT1 may have a unique
and important biological function. The sequence of this
domain is characterized by a high degree of hydrophobicity
and is highly conserved among different species [1].
Correspondence to H. Sun, Department of Chemistry, The University
of Hong Kong, Pokfulam Road, Hong Kong.
Fax: + 852 2857 1586, Tel.: + 852 2859 8974,
E-mail: hsun@hkucc.hku.hk
Abbreviations: doxylPtdCho, palmitol(doxyl) stearoyl-phosphatidyl-
choline; 12-doxylPtdCho, doxylPtdCho lipids containing the nitroxide
label on C12; DMT1, divalent metal transporter; DMT1-TM4,
transmembrane domain 4 of DMT1; HFIP, 1,1,1,3,3,3,-hexafluoro-2-
propanol; TFE, 2,2,2-trifluoroethanol; TM, transmembrane domain.
Note: The coordinate for the 16 lowest energy conformers both in
SDS micelles at pH 6.0 and TFE has been deposited in the protein data
bank (http://www.rcsb.org/pdb/index.html).
(Received 29 January 2004, revised 16 March 2004,
accepted 23 March 2004)
Eur. J. Biochem. 271, 1938–1951 (2004) FEBS 2004 doi:10.1111/j.1432-1033.2004.04104.x

Although numerous studies have been carried out to
explore the molecular biology aspects of DMT1 since the
discovery of this gene, there has so far been no structural
characterization of either this integral protein or a segment
of it. Analysis of the structure of membrane proteins either
by NMR spectroscopy or crystallography has proven
difficult, because the native structures of these integral
proteins are largely dependent on the associated membrane.
Recently model peptides, which mimic the sequence of a
segment or a subunit of membrane proteins, have been
widely used to investigate structure and function in several
integral membrane proteins [15–20]. This approach has
proved to be very successful in providing qualitative
structural information and in guiding complete structure
determination [21,22]. For example, it has enabled 3D
structural models of lactose permease, a 12-transmembrane
helix bundle that transduces free energy, to be derived
recently, based on its transmembrane topology, secondary
structure, and numerous interhelical contacts without using
crystals [23].
We have previously investigated the secondary structure
of the TM4 of DMT1 in various membrane-mimetic
environments, such as 2,2,2-trifluoroethanol (TFE), deter-
gent micelles and phosphate lipids [24]. We showed that the
DMT1-TM4 peptide assumed predominately an a-helical
conformation in these environments. In the present study,
we have used NMR spectroscopy and a molecular dynamic
simulated annealing approach to characterize the 3D
structures of DMT1-TM4 in both TFE and SDS micelles
at different pH values. The topology of the peptide in SDS
micelles was probed by the effects of spin-labels, including
both palmitol(doxyl) stearoyl-phosphatidyl-choline (doxy-
lPtdCho) lipids containing the nitroxide label on C12
(12-doxylPtdCho) and paramagnetic metal ions (Mn
2+
and
Gd
3+
), on the intensities of NMR signals. The peptide was
found to embed into the interior of SDS micelles. The
possibility of formation of a divalent metal channel has been
discussed.
Experimental procedures
Materials
The sequence of the peptide (RVPLYGGVLITIADT
FVFLFLDKY) was taken from rat DMT1 and represents
the putative TM4 (residues 179–202). The peptide was
synthesized by a solid-phase method and was purified by
HPLC on a Zorbax SB Phenyl reverse phase column using
0.1% (v/v) trifluoroacetic acid/water and 0.1% (v/v)
trifluoroacetic acid/acetonitrile as solvents (Biopeptide
Co., LLC. San Diego, CA, USA). The purity was assessed
by both mass spectrometry and analytical HPLC to be
above 95%. 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was
obtained from Sigma. Deuterated reagents for NMR
sample preparation, e.g. 2,2,2-trifluoroethanol-d
2
and
2,2,2,-trifluoroethanol-d
3
99.94% (TFE), methanol-d
4
99.6%, deuterium oxide 99.96%, and sodium dodecyl-d
25
sulfate were purchased from Cambridge Isotope Laborat-
ories (Cambridge, MA, USA). Palmitol(doxyl)-stearoyl-
phosphatidylcholine (doxylPtdCho) lipids containing the
nitroxide spin label on C12 were purchased from Avanti
Polar Lipids (Alabaster, AL, USA).
Circular dichroism spectroscopy
CD experiments were performed on a Jasco J-720 spectro-
polarimeter at ambient temperature. Cells with path lengths
of 0.1 and 1.0 mm were employed for sample solutions
containing final peptide concentrations of 6, 12, 23, 47, 94,
188, 375 and 750 l
M
in TFE. Spectra were recorded from
190 to 260 nm at a scan rate of 50 nmÆmin
)1
with a respond
time of 0.25 s, step resolution of 0.1 nm and band width of
1 nm. Each spectrum was obtained from the average of four
scans. Prior to calculation of final ellipiticity, all spectra were
corrected by subtraction of background and were smoothed
using a fast Fourier transform filter.
NMR spectroscopy
The samples used for NMR studies were prepared as
described previously [24]. Briefly, 3 mg of the peptide
dissolved in HFIP was mixed with an equal volume of
SDS-d
25
aqueous solution. The mixture was further diluted
with water, and was subject to lyophilization. The resulting
powder was then redissolved in 0.6 mL H
2
O containing
10% (v/v) D
2
O. The concentration of the peptide was
approximately 2.0 m
M
in SDS-d
25
(300 m
M
). Spectra for
assignments and structure calculation in the presence of
SDS were recorded at 298 K. In the presence of TFE, the
spectra were recorded at 298 and 305 K to resolve spectral
overlap.
All spectra were recorded on a Bruker AV600 spectro-
meter, operating at a proton frequency of 600.13 MHz.
Water suppression was carried out using a 3-9-19 watergate-
pulse sequence [25,26]. The sodium salt of trimethylsilyl-
propionate-d
4
solution was used to reference chemical shifts.
1D experiments were acquired using 32 768 data points and
processed with 0.3 Hz line broadening. The NOESY [27,28]
experiments were recorded at mixing times of 50, 150, 200
and 250 ms, and the TOCSY spectra employed the MLEV-
17 pulse sequence [29] with mixing periods of 50–100 ms.
The relaxation delay was 1.8 s in the TOCSY experiments
and 2.0 s in the NOESY experiments. Typically, 40–80
transients were collected for each increment of F1 in the
NOESY experiments, and 80–120 in the TOCSY experi-
ments. All 2D experiments were collected using 2048 data
points in F2, 256–512 increments in F1. All 2D Spectra were
acquired in the phase sensitive mode using States-time-
proportional phase incrementation in F1 dimension.
Spectral data were processed on a computer using
standard Bruker software (
XWINNMR
Version 3.1). Data
were zero-filled to 2048 points in F1 dimension and then
transformed with a shifted sine-bell squared window
function in both dimensions. Base line correction was also
carried out.
Structure calculations
Distance constraints were obtained from NOESY spectra
recorded with a mixing time of 200 ms in SDS micelles and
150 ms in TFE at 298 and 305 K, respectively. In the case
of severe spectral overlap, the corresponding NOEs were
excluded from the set used for the structure calculations.
Both NOE intensities and chemical shifts were extracted
using the
SPARKY
software [30] and served as an input for
FEBS 2004 Structure and topology of TM4 of DMT1 (Eur. J. Biochem. 271) 1939

the program of
CYANA
(1.0) [31]. On the basis of these
distance constraints obtained using the macro
CALIBA
,a
systematic analysis of the local conformation around the C
a
atom of each residue, including the dihedral angles /,w,v
1
and v
2
, was performed using the macro
GRIDSEARCH
as
implemented in
CYANA
[31]. The final nonredundant upper-
limit constraints and the resulting angle constraints were
used in the structural calculations. No stereospecific assign-
ments were obtained in any case. The 200 randomized
starting structures were energy minimized during 4000 steps
under the NMR constraints, and the 30
CYANA
conformers
with the lowest target function values were selected for
further energy minimization under the force field of Cornell
et al. [32] using a generalized Born solvent model with a
water shell of 8 A
˚in
AMBER
7 [33,34].
From these calculated structures, 16 conformers with the
lowest energy were selected to represent the NMR struc-
tures. The quality of the final structures was accessed using
the program of
PROCHECK
-
NMR
[35]. Further analysis and
visualization of the conformers including calculation of root
mean square deviations (rmsds) and identification of
H-bonds was performed using the molecular graphics
program
MOLMOL
[36].
Paramagnetic broadening experiments
Samples containing 2 m
M
DMT1-TM4 and 300 m
M
SDS-
d
25
in 0.6 mL 90% H
2
O/10% D
2
O (v/v) were used in these
experiments. The pH was adjusted to either 5.5 or 7.4 by
addition of small aliquots of NaOH. Spin-labeled 12-
doxylPtdCho was solubilized in methonal-d
4
, and aliquots
of this solution were then added to the peptide at pH 5.5 to
yield a final concentration of the spin-label of 5 m
M
.This
corresponded to approximately one spin-label per micelle
on the assumption of about 60 molecules per micelle [37].
The TOCSY spectrum (mixing time of 50 ms) was
acquired with a spectral width of 6 p.p.m. in F1 dimension,
with 120 transients, and 256 increments in F1 dimension.
The pH of the sample was then raised to 7.4 and the
TOCSY spectrum was collected. The reference spectrum in
the absence of the spin-label was also recorded under
identical conditions. For Gd
3+
and Mn
2+
broadening
studies, either GdCl
3
or MnCl
2
were dissolved in H
2
O
before being added to the sample. The experiments were
performed with concentrations of paramagnetic metal ions
of 0.1, 0.2, 0.4 and 1.0 m
M
. The TOCSY spectra were
again recorded in the presence of different amounts of
paramagnetic metal ions at different pH values (e.g. 7.4, 5.5
and 4.0).
Hydrogen exchange experiments
In the TFE system, 3 mg of DMT1-TM4 was directly
dissolved in 0.6 mL TFE-d
3
. Fast exchange amide protons
were monitored by subsequently recording a series of one-
dimensional
1
H-NMR spectra at 10, 30, 60, 90, 120 and
360 min until no further changes were observed in the
spectra. The TOCSY spectrum (mixing time 50 ms) was
then acquired in a total time of 19 h, and those protons
which showed cross-peaks in the H
a
–H
N
region of TOCSY
spectrum were regarded as slowly exchanging amide
protons.
In SDS micelles, 0.6 mL D
2
O was added to lyophilized
samples containing 2 m
M
peptide and 300 m
M
SDS-d
25
.
The pD was adjusted to 5.5 by addition of aliquots of
NaOD. Similarly, the fast exchanging amide protons were
monitored by 1D proton NMR spectra recorded at different
time intervals from 10 to 120 min. The NOESY spectrum
(mixing time 200 ms) was then acquired in a total time of
8.5 h, and the protons that appeared in the spectrum were
regarded as relatively slow exchanging protons. All amide
protons were exchanged completely within 12 h.
Results
Resonance assignment and secondary structure
determination
The DMT1-TM4 peptide is highly hydrophobic, and insol-
uble in water and a range of organic solvents. We have chosen
TFE and SDS to solubilize the peptide and to mimic
biological membranes. The peptide is stable in these environ-
ments for at least a couple of months at room temperature.
TOCSY and NOESY spectra with a set of mixing times were
recorded for DMT1-TM4 in SDS-d
25
at different pH values,
and reasonably well-resolved spectra were found at a wide
range of pH values. The spectra recorded in SDS micelles at
pH 6.0 were chosen for sequential assignments and structural
calculations, as it is close to the biological function pH (5.5)
of its integral protein. Moreover, the spectra at this pH were
relatively well resolved compared with those at other pH
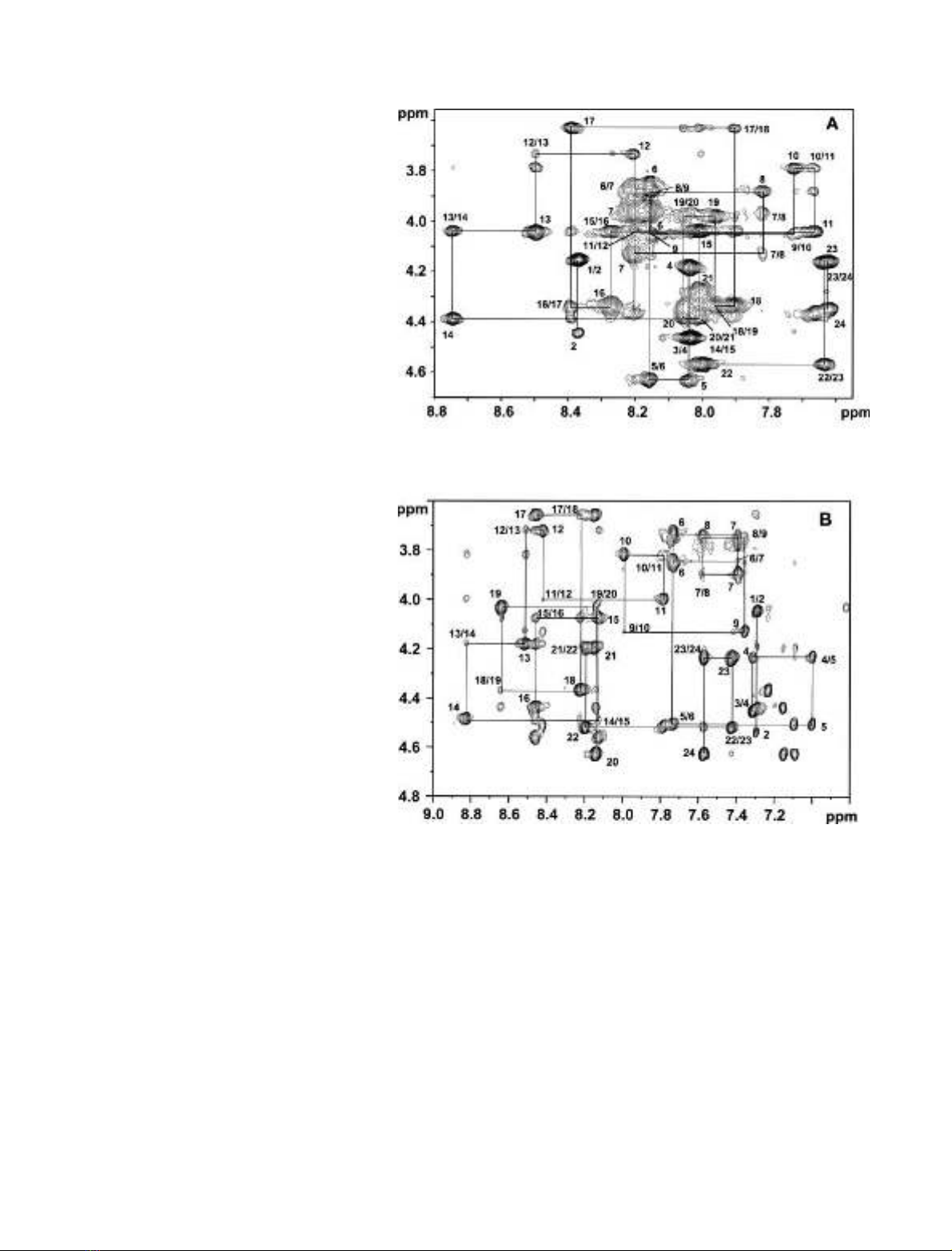
values. Figure 1 shows the fingerprint region of the 600 MHz
NOESY spectra of DMT1-TM4 in 300 m
M
SDS-d
25
at
pH 6.0 (298 K) and in TFE-d
2
(305 K). It can be seen that the
peptide exhibited sufficient chemical shift dispersions in both
environments, allowing unambiguous assignments of most
proton frequencies. The
1
H resonance assignment was
straightforward, based on a standard procedure [38]. The
complete spin systems of the individual amino acid residues
were identified using the TOCSY spectra with mixing times of
50 and 100 ms. The backbone sequential connectivities were
established by following the H
a
and H
N
cross-peaks of
adjacent amino acids in the fingerprint and the H
N
–H
N
region
of the TOCSY and NOESY spectra. Using this technique it
was possible to unambiguously assign almost all the proton
resonances including side chains, apart from a few aromatic
protons from phenylalanine residues due to spectral overlap
(H. Li, F. Li, Z. M. Qian & H. Sun, unpublished observation).
We noticed that chemical shift values in TFE and in SDS
micelles were similar with the expected exception of amide
protons, in particular, the amide protons of N-terminal
residues.
The chemical shifts of the H
a
protons provide informa-
tion about secondary structural elements of the peptides.
Generally, all residues experience a H
a
upfield shift relative
to the random-coil value when adopting a helical confor-
mation and a downfield shift when found in an extended or
b-strand structure [39]. The peptide was predicted to adopt a
helical conformation in the segment of Leu9–Phe20 in SDS
micelles and Gly6–Lys23 in TFE from the chemical shift
index method [40] (Fig. 2). Further investigation by exam-
ining the observed NOE connectivities produced similar
results. All H
N
resonances from Tyr5 to Phe20 were found
to be connected by (i, i+1) connectivities except for Phe16
1940 H. Li et al.(Eur. J. Biochem. 271)FEBS 2004
and Val17, which are overlapped together in TFE (Fig. 2),
indicative of a helical conformation in this region. This is
in agreement with our previous CD studies, which demon-
strated high helical contents in the DMT1-TM4 peptide
[24]. Furthermore, we also observed that the chemical shift
for threonine H
b
is greater than that of H
a
for both Thr11
and Thr15, indicating that both threonines are situated in
the helical region. Evaluation of the secondary structure
from backbone coupling constants was hampered due to
extensive line broadening both in the TFE and SDS micelle
environments, which retards determination of these coup-
ling constants.
Structure calculations and description
Distance constraints were obtained from NOESY spectra
recorded with a mixing time of 200 ms measured in 90%
H
2
O/10% D
2
O (v/v) containing 2 m
M
peptide and 300 m
M
SDS-d
25
at pH 6.0, and 150 ms in TFE-d
2
.TheNOE
connectivities and numbers of NOEs per constraints for
DMT1-TM4 in both solvents are summarized in Fig. 2.
Except for unresolved cross-peaks between the residue pairs
Leu21/Asp22 in SDS, and Phe16/Val17 and Phe20/Leu21 in
TFE, almost all of the possible HN
i=HN
iþ1[38], and sequential
NOEs were observed in the segment of Tyr5–Phe20. In
addition, the presence of medium-range connectivities [38],
such as Ha
i=HN
iþ3,H
a
i=Hb
iþ3and Ha
i=HN
iþ4was also observed
for Val8–Phe20 in SDS and Val8–Lys23 in TFE, indicative
of a well-structured peptide in helical conformation over
each span [41,42]. The absence of medium-range NOEs
at the N-terminus suggested no defined structure in this
segment. However, in the C-terminal segment, NOEs
between Ha
i=HN
iþ2and HN
i=HN
iþ2, which are characteristic
of 3
10
-helix [38], were also detected in TFE. No long-range
NOEs were observed over the full peptide, indicating that
the peptide does not form tertiary folds.
Fig. 1. Fingerprint region of the 600 MHz
NOESY spectra of DMT1-TM4. (A) 200 ms
NOE spectrum of 2 m
M
DMT1-TM4 in
300 m
M
SDS-d
25
at pH 6.0, 298 K. (B) 150 ms
NOE spectrum of 2 m
M
peptide in TFE-d
2
,
305 K. The sequential assignment of all resi-
dues is indicated.
FEBS 2004 Structure and topology of TM4 of DMT1 (Eur. J. Biochem. 271) 1941

Totals of 241 and 265 meaningful upper-limit distance
constraints were obtained based on totals of 358 and 378
assigned NOE cross-peaks for DMT1-TM4 in SDS at
pH 6.0 and in TFE, respectively. A total of 79 dihedral
angle constraints for 50 angles in SDS vs. 85 constraints for
55 angles in TFE, derived using the macro
GRIDSEARCH
as
implemented in
CYANA
[31], were also included in the
structure calculations. In no case could stereospecific
assignment be achieved. The structures were calculated by
molecular dynamics in torsion angle space using a simulated
annealing protocol as implemented in the program
CYANA
[31]. Under this protocol, 200 randomized starting struc-
tures were energy minimized under the NMR constraints
and the 30 structures with no violations > 0.2 A
˚for the
distance constraint and > 5for the angle constraint, as well
as with the lowest target function were selected in either
SDS or TFE for further energy minimization. The structural
statistics showed that the structures of DMT1-TM4 in both
membrane-mimetic environments were well defined by
NMR data, as indicated by the low values of the target
function (Table 1). The backbone /and wdihedral angles
were also uniformly well-defined, as judged from an angular
order parameter of 1.0 in the span of Leu9–Phe20 [43].
These structures were subjected to an energy minimization
using the program
AMBER
7 [33,34] in the
AMBER
force field
[32]. The final 16 lowest energy structures of DMT1-TM4 in
both SDS (pH 6.0) and TFE were chosen to represent the
solution structures of the peptide, as shown in Fig. 3.
The quality of the final structures was assessed using the
program
PROCHECK
-
NMR
[35]. In the range of well-defined
residues, i.e. Leu9–Phe18 in SDS (pH 6.0) and Leu9–Phe20
in TFE, 99.4% and 91.7% occupy the most favored regions
of the Ramachandran space in SDS and TFE, respectively,
and none are found in the disallowed regions (Table 1).
The overall structure of DMT1-TM4 in SDS micelles is
similar to that in TFE. The mean structures obtained from
MOLMOL
showed that the peptide folded into an a-helical
conformation for Leu9–Phe18 in SDS and Leu9–Phe20 in
TFE. The pairwise rmsds between the minimized structures
and the mean structure in SDS at pH 6.0 were 0.18 ± 0.06
and 0.85 ± 0.17 A
˚for the backbone and all heavy atoms,
respectively, in the segment Leu9–Phe18, vs. 0.20 ± 0.06
and 0.89 ± 0.16 A
˚in the segment Leu9–Phe20 (Table 1).
The pairwise rmsds between the minimized structures
and mean structure in TFE were 0.23 ± 0.10 and
0.82 ± 0.17 A
˚for the backbone and all heavy atoms,
respectively, in the segment Leu9–Phe20, vs. 0.26 ± 0.12
and 0.87 ± 0.17 A
˚in the segment Leu9–Lys23. This
suggested that the structures of the peptide from the residue
Leu9 towards the C-terminal residues were well-defined by
NMR constraints, which is consistent with the observed
pattern of sequential and medium-range NOEs and the
Fig. 2. Summary of NMR spectroscopy data for secondary structure prediction for DMT1-TM4 peptide. (A) In SDS micelles at pH 6.0, 298 K and
(B) in TFE at 305 K. The NOE connectivities, amide proton exchange rates, chemical shift index values as well as numbers of NOE constraints per
residue for DMT1-TM4 are shown. Slowly and rapidly exchanging amide protons are represented as filled and open circles, respectively. The NOEs
of intra, sequential and medium range are indicated as white, light gray and dark gray bars, respectively.
1942 H. Li et al.(Eur. J. Biochem. 271)FEBS 2004

























