
An active triple-catalytic hybrid enzyme engineered
by linking cyclo-oxygenase isoform-1 to prostacyclin
synthase that can constantly biosynthesize prostacyclin,
the vascular protector
Ke-He Ruan, Shui-Ping So, Vanessa Cervantes, Hanjing Wu*, Cori Wijaya and Rebecca R. Jentzen*
Department of Pharmacological and Pharmaceutical Sciences, Center for Experimental Therapeutics and PharmacoInformatics,
University of Houston, TX, USA
Prostacyclin (prostaglandin I
2
, PGI
2
) [1], which has
strong antiplatelet aggregation and vasodilation prop-
erties [1–4], and is synthesized from endothelial and
vascular smooth muscle cells, has been identified as
one of the most important vascular protectors against
thrombosis and heart disease [5]. Recently, there have
been many new studies that have confirmed the impor-
tance of PGI
2
in vascular protection. For instance, it
Keywords
COX; cyclo-oxygenase; PG12; prostacyclin;
prostaglandin 12
Correspondence
K.-H. Ruan, Department of Pharmacological
and Pharmaceutical Sciences, Center for
Experimental Therapeutics and
PharmacoInformatics, University of
Houston, Room 521, Science & Research 2
Building, Houston, TX 77204-5037, USA
Fax: +1 713 743 1884
Tel: +1 713 743 1771
E-mail: khruan@uh.edu
*Present address
The University of Texas Health Science
Center, Houston, TX, USA
(Received 15 July 2008,
revised 23 September 2008,
accepted 25 September 2008)
doi:10.1111/j.1742-4658.2008.06703.x
It remains a challenge to achieve the stable and long-term expression (in
human cell lines) of a previously engineered hybrid enzyme [triple-catalytic
(Trip-cat) enzyme-2; Ruan KH, Deng H & So SP (2006) Biochemistry 45,
14003–14011], which links cyclo-oxygenase isoform-2 (COX-2) to prostacy-
clin (PGI
2
) synthase (PGIS) for the direct conversion of arachidonic acid
into PGI
2
through the enzyme’s Trip-cat functions. The stable upregulation
of the biosynthesis of the vascular protector, PGI
2
, in cells is an ideal
model for the prevention and treatment of thromboxane A
2
(TXA
2
)-medi-
ated thrombosis and vasoconstriction, both of which cause stroke, myo-
cardial infarction, and hypertension. Here, we report another case of
engineering of the Trip-cat enzyme, in which human cyclo-oxygenase iso-
form-1, which has a different C-terminal sequence from COX-2, was linked
to PGI
2
synthase and called Trip-cat enzyme-1. Transient expression of
recombinant Trip-cat enzyme-1 in HEK293 cells led to 3–5-fold higher
expression capacity and better PGI
2
-synthesizing activity as compared to
that of the previously engineered Trip-cat enzyme-2. Furthermore, an
HEK293 cell line that can stably express the active new Trip-cat enzyme-1
and constantly synthesize the bioactive PGI
2
was established by a screening
approach. In addition, the stable HEK293 cell line, with constant produc-
tion of PGI
2
, revealed strong antiplatelet aggregation properties through its
unique dual functions (increasing PGI
2
production while decreasing TXA
2
production) in TXA
2
synthase-rich plasma. This study has optimized engi-
neering of the active Trip-cat enzyme, allowing it to become the first to
stably upregulate PGI
2
biosynthesis in a human cell line, which provides a
basis for developing a PGI
2
-producing therapeutic cell line for use against
vascular diseases.
Abbreviations
AA, arachidonic acid; COX, cyclo-oxygenase; COX-1, cyclo-oxygenase isoform-1; COX-2, cyclo-oxygenase isoform-2; ER, endoplasmic
reticulum; FITC, fluorescein isothiocyanate; IP
,
PGI
2
receptor; PGE
2
, prostaglandin E
2
; PGF
2
, prostaglandin F
2
; PGG
2,
prostaglandin G
2;
PGH
2,
prostaglandin H
2;
PGI
2,
prostaglandin I
2
(prostacyclin); PGIS, prostaglandin I
2
(prostacyclin) synthase; SLO, streptolysin-O; TM,
transmembrane domain; TXA
2,
thromboxane A
2;
TXAS, thromboxane A
2
synthase.
5820 FEBS Journal 275 (2008) 5820–5829 ª2008 The Authors Journal compilation ª2008 FEBS
was discovered that PGI
2
receptor (IP) -knockout mice
showed an increase in thrombosis tendency [6]. Also,
the suppression of PGI
2
biosynthesis by cyclo-oxygen-
ase isoform-2 (COX-2) inhibitors was linked to
increased rates of heart disease in human clinical trials
[7]. Thus, increasing the biosynthesis of PGI
2
would be
very useful for protection of the vascular system. It is
known that the biosynthesis of prostanoids through
the arachidonate– cyclo-oxygenase (COX) pathway
occurs when arachidonic acid (AA) is first converted
into prostaglandin G
2
(PGG
2
, catalytic step 1), and
then to prostaglandin endoperoxide [prostaglandin H
2
(PGH
2
)] (catalytic step 2) by COX isoform-1 (COX-1)
or COX-2 in cells [8]. The PGH
2
then serves as a com-
mon substrate for downstream synthases, and is isom-
erized to prostaglandin D
2
, prostaglandin E
2
(PGE
2
),
prostaglandin F
2
(PGF
2
), and prostaglandin I
2
(PGI
2
)
or thromboxane A
2
(TXA
2
) by individual synthases
(catalytic step 3). The overproduction of TXA
2
, a pro-
aggregatory and vasoconstricting mediator, has been
identified as one of the key factors causing thrombosis,
stroke, and heart disease [1,2]. PGI
2
is the primary AA
metabolite in vascular walls, and has opposite biolo-
gical properties to that of TXA
2
; it therefore represents
the most potent endogenous vascular protector, acting
as an inhibitor of platelet aggregation and a strong
vasodilator on vascular beds [9–12]. Specifically
increasing PGI
2
biosynthesis requires a highly efficient
chain reaction between COX and PGI
2
synthase
(PGIS), which consists of triple catalytic (Trip-cat)
functions.
Recently, we engineered a hybrid enzymatic protein
with the ability to perform the Trip-cat functions by
linking the inducible COX-2 to PGIS through a trans-
membrane (TM) domain [13,14]. Here, we refer to this
previously engineered enzyme as Trip-cat enzyme-2.
Transient expression of active Trip-cat enzyme-2 in
HEK293 and COS-7 cells has been demonstrated.
However, there are concerns in using Trip-cat enzyme-
2in vivo, because COX-2 has an inducible nature, has
a lower capacity to be stably expressed, and may also
lead to numerous pathological processes, such as
cancers and inflammation. Given the nature of COX-1,
a housekeeping enzyme that is consistently expressed
in cells, we hypothesize that a Trip-cat enzyme,
constructed by linking COX-1 to PGIS, is likely to
demonstrate stable expression in cells and therefore
lead to constant production of the vascular protective
prostanoid PGI
2
. To test this hypothesis, in this article
we report the construction of a new Trip-cat enzyme
linking COX-1 to PGIS, which we call Trip-cat
enzyme-1. Our studies have confirmed that Trip-cat
enzyme-1 can be stably expressed in HEK293 cells and
therefore lead to the generation of a cell line that con-
stantly delivers the vascular protector PGI
2
. This study
has provided a fundamental step towards specifically
and stably upregulating PGI
2
biosynthesis in thera-
peutic cells for the prevention and treatment of throm-
bosis and heart disease.
Results
Design of a new-generation Trip-cat enzyme
(COX-1 linked to PGIS) that directly converts
AA to the vascular protector PGI
2
As described above, we recently invented an approach
for engineering an active hybrid enzyme (Trip-cat
enzyme-2), by linking human COX-2 to PGIS (COX-2–
linker–PGIS), which demonstrated Trip-cat activities in
converting AA to PGG
2
, PGH
2
, and finally PGI
2
[13,14]
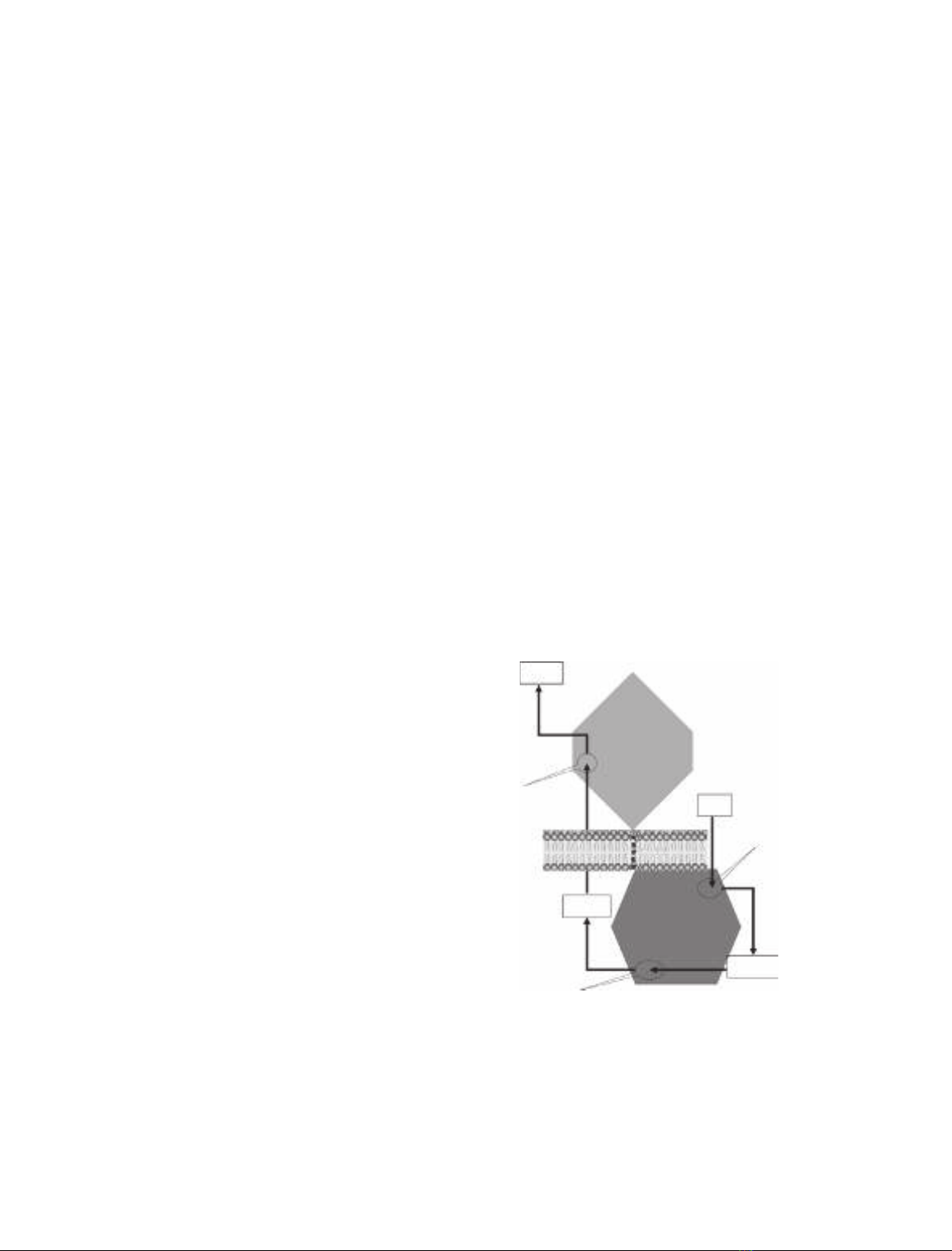
(Fig. 1). This finding provided great potential for specif-
ically upregulating PGI
2
biosynthesis in ischemic tissues
through the introduction of the Trip-cat enzyme-1 gene
into these target tissues. On the other hand, there is the
COX-1 enzyme, which is well known to have a similar
function (coupling to PGIS to synthesize PGI
2
in vitro
and in vivo) to that of COX-2. The housekeeping
enzyme COX-1, which has less pathological impact,
could be safer for gene and cell therapies than COX-2,
which is involved in the pathological processes of
PGI2
PGH2
PGG2
3rd
Catalytic
reaction
1st
Catalytic
reaction
2nd
Catalytic
reaction
PGIS
Substrate
AA
TM linker
COX-1
Fig. 1. A model of the newly designed Trip-cat enzyme-1. Trip-cat
enzyme-1 was created by linking COX-1 to PGIS through an opti-
mized TM linker (10 amino acid residues) without alteration of the
protein topologies in the ER membrane. The three catalytic sites in
and reaction products of COX-1 and PGIS are shown.
K.-H. Ruan et al. Prostacyclin-synthesizing protein with COX-1 and PGIS properties
FEBS Journal 275 (2008) 5820–5829 ª2008 The Authors Journal compilation ª2008 FEBS 5821

inflammation and cancers, and shows inducible tran-
sient expression. This suggested that the Trip-cat
enzyme containing COX-1 (Fig. 1) may have better
therapeutic potential than that containing COX-2 in
terms of stable expression in cells and pathogenic prop-
erties. Also, the X-ray crystal structure shows that the
membrane orientation and the membrane anchor
domain of COX-1 are similar to those of COX-2. This
led us to design a single molecule containing the cDNA
of human COX-1 and PGIS with a connecting TM lin-
ker derived from human bovine rhodopsin [15] (Fig. 1).
Cloning of Trip-cat enzyme-1 by linking COX-1
to PGIS
A PCR approach was used to link the C-terminus of
human COX-1 (NCBI GenBank ID: NM_080591) to
human PGIS (NCBI GenBank ID: D38145) by a heli-
cal linker with 10 residues (His-Ala-Ile-Met-Gly-
Val-Ala-Phe-Thr-Trp) derived from human rhodopsin.
The resultant cDNA sequence encoding the novel
Trip-cat enzyme-1 (COX-1–10aa–PGIS) was then sub-
cloned into the pcDNA3.1 vector for mammalian cell
expression [13]. Note that the entire cDNA sequence
of Trip-cat enzyme-1 encodes a single human protein
sequence, which could be used for therapeutics.
Expression of the engineered Trip-cat enzyme-1
in HEK293 cells
Despite the many similarities between human COX-1
and COX-2, there are several important differences.
For example, it has been reported that the C-terminal
Leu and the last six residues of COX-1 are important
for the enzyme’s activity [16]. However, they are not
identical to those of COX-2. Therefore, it was interest-
ing to investigate whether the linkage (from the C-ter-
minal Leu of COX-1 to the N-terminus of PGIS) in
Trip-cat enzyme-1 would affect its expression, protein
folding, and enzyme activity. Using the constructed
pcDNA3.1 COX-1–10aa–PGIS plasmid, the recombi-
nant COX-1–10aa–PGIS protein was successfully over-
expressed in the HEK293 cell line, showing the correct
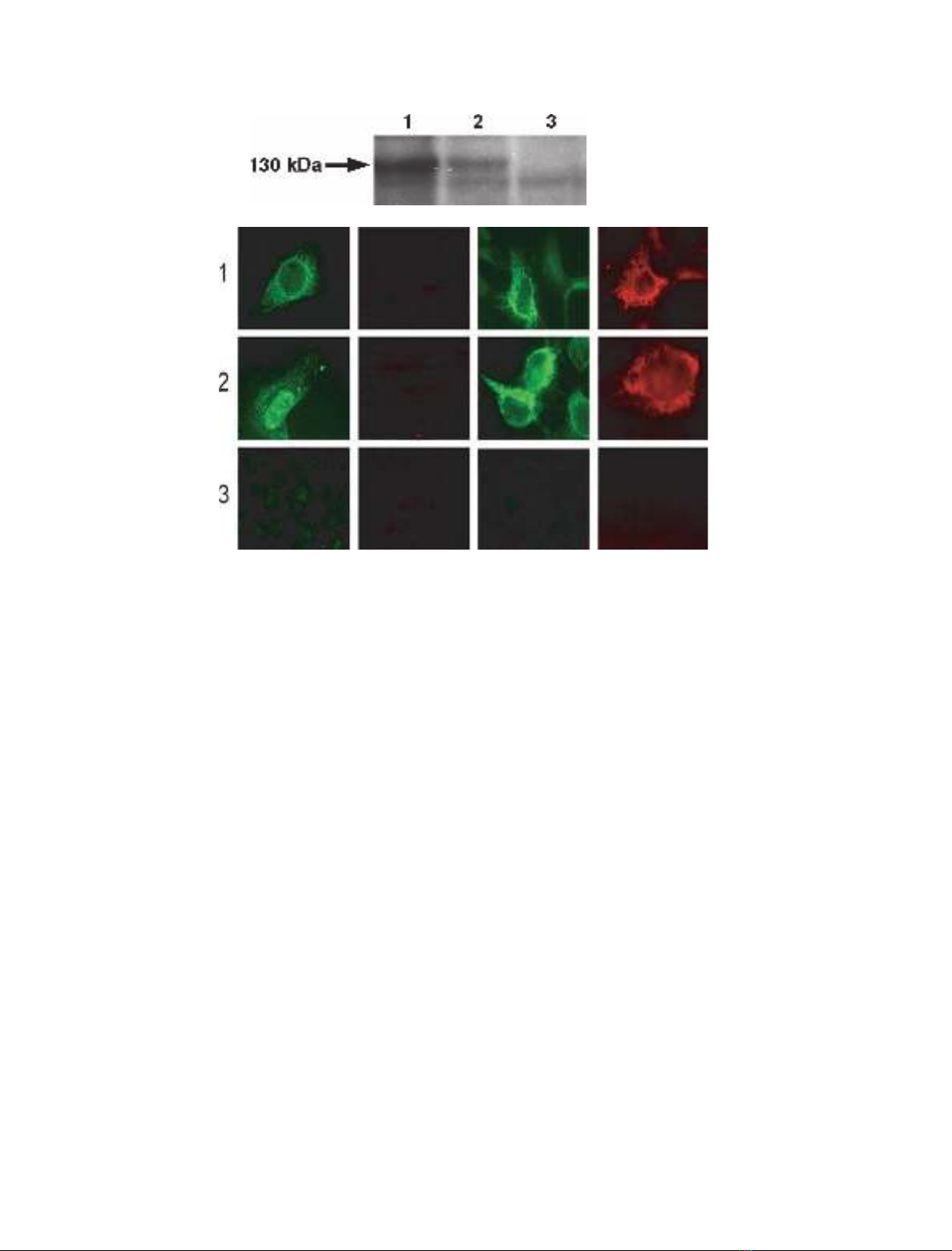
molecular mass of approximately 130 kDa in western
blot analysis (Fig. 2A, lane 1). This indicated that the
linkage from the C-terminal Leu of COX-1 to the
N-terminus of PGIS had no effect on Trip-cat enzyme
expression. In addition, a comparison of the expression
levels between COX-1–10aa–PGIS and COX-2–10aa–
PGIS revealed that the transfected HEK293 cells
expressed approximately three-fold more COX-1–
10aa–PGIS protein than COX-2–10aa–PGIS protein
under identical conditions (Fig. 2A, lane 2).
Subcellular localization of COX-1–10aa–PGIS
To determine whether the linkage of the C-terminal
Leu of COX-1 to PGIS had any effects on the sub-
cellular localization of Trip-cat enzyme-1, HEK293
cells expressing the enzyme COX-1–10aa–PGIS were
permeabilized and stained. Nonsignificant differences
were observed in the endoplasmic reticulum (ER)
staining patterns for the cells treated with streptolysin-
O (SLO), which selectively permeabilized the cell
membrane, and with saponin, which generally permea-
bilized both the cell and the ER membranes (Fig. 2B).
The results indicated that the modification of the link-
age between the COX-1 Leu residue and the PGIS
N-terminus had no significant effect on the subcellular
localization of COX-1–10aa–PGIS in the cells. The
idea that the PGIS domain is located on the cytoplas-
mic side of the ER and that the COX-1 domain is
located on the ER lumen for the overexpressed COX-
1–10aa–PGIS was also supported by immunostaining.
Antibody against PGIS was used to stain the cells trea-
ted with SLO or saponin, but antibody against COX-1
would only stain the cells treated with saponin
(Fig. 2B). These data further confirmed that the 10
amino acid linkage between COX-1 to PGIS had no
significant effects on the subcellular localization of
COX-1 and PGIS in the ER membrane.
Trip-cat activities of Trip-cat enzyme-1 in directly
converting AA to the vascular protector PGI
2
The biological activities of HEK293 cells expressing
the different eicosanoid-synthesizing enzymes that con-
vert AA to PGI
2
were assayed by the addition of
[
14
C]AA. The resultant [
14
C]eicosanoids, metabolized
by the enzymes in the cells, were profiled by HPLC
analysis (HPLC separation linked to an automatic
scintillation analyzer; Fig. 3). The Trip-cat activities
that occur during the conversion of [
14
C]AA to [
14
C]6-
keto-PGF
1a
(degraded PGI
2
) require two individual
enzymes, COX-1 and PGIS, in HEK293 cells
(Fig. 3A), because neither COX-1 (Fig. 3B) nor PGIS
(Fig. 3C) alone could produce [
14
C]6-keto-PGF
1a
from
[
14
C]AA in HEK293 cells. However, the cells express-
ing Trip-cat enzyme-1 were able to integrate the Trip-
cat activities of COX-1 and PGIS by converting the
added [
14
C]AA to the end-product, [
14
C]6-keto-PGF
1a
(Fig. 3D). It should be noted that in HEK293 cells
expressing Trip-cat enzyme-1, most of the added
[
14
C]AA was converted to [
14
C]6-keto-PGF
1a
, with
very low amounts of byproducts. In contrast, the cells
coexpressing COX-1 and PGIS synthesized less PGI
2
and produced significant amounts of other unidentified
Prostacyclin-synthesizing protein with COX-1 and PGIS properties K.-H. Ruan et al.
5822 FEBS Journal 275 (2008) 5820–5829 ª2008 The Authors Journal compilation ª2008 FEBS
lipid molecules. These data clearly indicated that the
enzymatic conversion of AA to PGI
2
is more efficient
with Trip-cat enzyme-1 than with coexpressed individ-
ual COX-1 and PGIS.
Enzyme kinetics of Trip-cat enzyme-1 compared
to those of its parent enzymes
In cells coexpressing COX-1 and PGIS, the coordina-
tion of COX-1 and PGIS in the ER membrane (for
the biosynthesis of PGI
2
from AA) is very fast. Only
120 s were required for 50% of the maximum activity
to be reached (Fig. 4A, triangles). The reaction was
almost saturated after approximately 5 min. The
amount of PGI
2
produced when the reaction was
extended from 5 min to 15 min increased by only 5%.
On the other hand, cells expressing the engineered
Trip-cat enzyme-1 (Fig. 4A, closed circles) showed the
same time-course pattern as that of the coexpressed
wild-type COX-1 and PGIS. In addition, Trip-cat
enzyme-1 also showed an identical dose-dependent
response to that of the parent enzymes in the biosyn-
thesis of PGI
2
(Fig. 4B). The K
m
and V
max
values for
Trip-cat enzyme-1 were approximately 5 and 400 lm,
respectively; these are almost identical to those of the
coexpressed COX-1 and PGIS. This study has indi-
cated that the expressed Trip-cat enzyme-1 in the cells
has correct protein folding, subcellular localization and
native enzymatic functions in a single folded protein,
similar to to its parent enzymes.
Establishing stable expression of Trip-cat
enzyme-1 in cells
Stable expression of the engineered Trip-cat enzyme-1
in cells is the basis for having the cells constantly pro-
duce PGI
2
. In this study, an HEK293 cell line was
used as the model for testing. After G418 screening for
b
acd
B
A
Fig. 2. (A) Western blot analysis for overexpressed COX-1–10aa–PGIS and COX-2–10aa–PGIS in HEK293 cells. HEK293 cells transiently trans-
fected with cDNA of COX-1–10aa–PGIS (lane 1) or COX-2–10aa–PGIS (lane 2), or the pcDNA3.1 vector alone (lane 3), were solubilized and
separated by 7% SDS ⁄PAGE, and then transferred to a nitrocellulose membrane. The expressed Trip-cat enzymes were stained with antibody
against PGIS. The molecular mass (130 kDa) of the engineered enzymes is indicated by an arrow. (B) Immunofluorescence micrographs of
HEK293 cells. In brief, the cells were grown on coverslides and transfected with the cDNA plasmid(s) of COX-1–10aa–PGIS (row 1), cotrans-
fected COX-1 and PGIS (row 2), or transfected with the pcDNA3.1 vector alone (row 3). The cells were permeabilized by SLO (columns a and
b) or saponin (columns c and d), and then incubated with affinity-purified rabbit antibody against PGIS peptide (columns a and c) or mouse
antibody against COX-1 (columns b and d) [13]. The bound antibodies were stained with FITC-labeled goat anti-(rabbit IgG) (columns a and c)
or rhodamine-labeled goat anti-(mouse IgG) (columns b and d). The stained cells were then examined by fluorescence microscopy [13].
K.-H. Ruan et al. Prostacyclin-synthesizing protein with COX-1 and PGIS properties
FEBS Journal 275 (2008) 5820–5829 ª2008 The Authors Journal compilation ª2008 FEBS 5823

the transiently transfected HEK293 cells containing the
Trip-cat enzyme-1 cDNA, cells stably expressing Trip-
cat enzyme-1 were successfully created, as indicated by
the enzyme activity assays showing continuous
[
14
C]PGI
2
production after the addition of [
14
C]AA
(Fig. 5, black squares). However, the same cells trans-
fected with COX-2–10aa–PGIS cDNA could only pro-
duce PGI
2
for a few days (Fig. 5, open squares), due
to a failure in the stable expression of Trip-cat
enzyme-2. This study indicated that the engineered
Trip-cat enzyme-1 most likely adopted the housekeep-
ing properties of COX-1, which produced constant
expression in the cells, whereas Trip-cat enzyme-2
mainly adopted the properties of inducible COX-2,
which expressed the protein for only a short period of
time.
Antiplatelet aggregation
The effects of HEK293 cells expressing COX-1–10aa–
PGIS on antiplatelet aggregation were explored. It is
known that platelets contain large amounts of COX-1
and thromboxane A
2
synthase (TXAS). When AA was
added to the platelet-rich plasma, the platelets began
to aggregate in minutes (Fig. 6A, line a). However, this
aggregation was completely blocked in the presence of
cells expressing COX-1–10aa–PGIS (Fig. 6A, line b).
In contrast, the aggregation was only partially blocked
in the presence of cells coexpressing COX-1 and PGIS
(Fig. 6A, line c). This indicated that AA was not only
converted into PGI
2
(by COX-1 and PGIS), to act
against platelet aggregation, but also converted into
TXA
2
, promoting platelet aggregation by the abundant
TXAS in the platelets. In contrast, no effects were
observed with the nontransfected, control HEK293
cells (Fig. 6A, line d). From these observations, it is
clear that the engineered Trip-cat enzyme-1 has supe-
rior antiplatelet aggregation activity to coexpressed
COX-1 and PGIS.
To test whether Trip-cat enzyme-1 can indirectly
inhibit platelet aggregation induced by other factors,
such as collagen (through non-COX pathways), it is
necessary to compare the effects of HEK293 cells
(expressing Trip-cat enzyme-1) on human platelets
induced by collagen (Fig. 6B, bars 1 and 2) with those
of the AA-induced platelets (Fig. 6B, bars 3 and 4). It
is clear that cells expressing Trip-cat enzyme-1 could
not only directly inhibit AA-induced platelet aggre-
gation (Fig. 6B, bar 4), but also significantly inhibit
collagen-induced platelet aggregation by up to 50%
(Fig. 6B, bar 2).
Competitively upregulating PGI
2
biosynthesis in
the presence of platelets
To further demonstrate the competitive upregulation
of PGI
2
biosynthesis by COX-1–10aa–PGIS in the
presence of TXAS, [
14
C]AA was added to platelet-rich
plasma containing endogenous COX-1 and TXAS, in
the presence and absence of cells stably expressing
CPM
0
100
200
300
400
A
[14C]-6-keto-PGF1α
[
14
C]-AA
010203040
0
100
200
300
400
D
[14C]-6-keto-PGF1α
[14C]-AA
0 10 20 30 40
0
100
200
300
400
C
[14C]-AA
0
100
200
300
400
B
Non specific peak
[
14
C]-AA
Fig. 3. Determination of the Trip-cat activi-
ties of the fusion enzymes for directly con-
verting AA to PGI
2
, using an isotope-HPLC
method for HEK293 cells. Briefly, the cells
(0.1 ·10
6
) transfected with the cDNA(s)
of both COX-1 and PGIS (A), COX-1 (B),
PGIS (C) and COX-1–10aa–PGIS (D) were
washed and then incubated with [
14
C]AA
(10 lM) for 5 min. The metabolized
[
14
C] eicosanoids produced from the [
14
C]AA
in the supernatant were analyzed by HPLC
on a C18 column (4.5 ·250 mm) connected
to a liquid scintillation analyzer. The total
counts for the specific peaks in each assay
are approximately: 400 counts in (A); 550
counts in (B); 600 counts in (C); and 750
counts in (D).
Prostacyclin-synthesizing protein with COX-1 and PGIS properties K.-H. Ruan et al.
5824 FEBS Journal 275 (2008) 5820–5829 ª2008 The Authors Journal compilation ª2008 FEBS

























