PHƢƠNG PHÁP VÀ KỸ THUẬT CƠ BẢN TRONG SHPT
GV: Nguyễn Thị Ngọc Yến
Mục tiêu
Công cụ cơ bản của SHPT và cách sử dụng Một số phương pháp cơ bản của SHPT Ứng dụng trong thực tế
Các enzym biến đổi acid nucleic
Enzym cắt hạn chế - RE
Restriction enzyme (RE): Endonuclease cắt ADN tại
hay gần 1 trình tự nhận diện đặc hiệu
ngăn cản tác động RE
Methylase: methyl hóa trình tự nhận diện đặc hiệu và
Cặp RE/methylase khác nhau
Phân loại RE
Loại II chỉ có hoạt tính endonuclease, không cần ATP để hoạt động và cắt ADN tại hoặc rất gần vị trí nhận diện sử dụng nhiều o EcoRII o BamHI…
Loại I và III là phức hệ gồm cả 2 hoạt tính RE và methyl hóa, loại này cần ATP và nó cắt ADN tại vị trí khá xa điểm nhận diện ít được dùng
Nhận diện vị trí cắt
Vị
trí nhận diện của đa số RE loại
II
thường chứa 4 -6
nucleotid theo kiểu palindrome
RE chỉ cắt ADN sợi đôi và tạo ra 2 đầu tận 5’-P và 3-OH. Đầu
cắt có thể tà hoặc so le (đầu dính)
Nếu trình tự nhận diện có phần nào tính đối xứng thì điểm cắt
thường nằm giữa nó
Polymerase – Đặc tính chung
Hoạt tính tổng hợp 5’3’: gắn dNTP vào đầu 3’-OH
của mồi và giải phóng PPi
Hoạt tính exonuclease 3’5’: “sửa lỗi” Hoạt tính exonuclease 5’3’: hủy mồi Ribonuclease H: hủy ARN trong phức lai ARN-ADN Dịch chuyển sợi Khả năng xử lý Tần suất lỗi
Polymerase – Đặc tính chung
Phân loại
ADN polymerase phụ thuộc ADN: enzym sao chép ARN polymerase phụ thuộc ADN: enzym phiên mã ADN polymerase phụthuộc ARN: reverse transcriptase
o Hoạt động tối ưu ở 75-80oC, giảm 10 lần ở 37oC. Hoạt tính
vẫn còn sau 2h ở 95oC
o Ứng dụng:
• Khuếch đại ADN bằng PCR
• Giải trình tự ADN bằng pp dideoxy
Taq polymerase
Ligase
Enzym xúc tác phản ứng tạo liên kết phosphodiester giữa 3’-OH và 5’-P của hai sợi acid nucleic
Chủng VSV và vector tạo dòng
Chủng vi sinh vật
E.coli Bộ máy di truyền được nghiên cứu đầy đủ Tốc độ tăng trưởng nhanh Khả năng gây bệnh thấp
Chủng vi sinh vật
o Nhân bản vector phage M13 o Dùng với plasmid pUC, plasmid sử dụng kỹ thuật bổ sung
anpha để chọn lọc dòng tái tổ hợp
o Mang gen cho đoạn omega của β-galactosidase trên plasmid F’, cho phép nó bị nhiễm bởi các phage dạng sợi như M13. Nếu mất plasmid F’ sẽ làm mất khả năng này
JM103
o Mang đặc tính bổ sung anpha như JM103 o Đột biến gen recA làm mất khả năng tái tổ hợp tương
đồng
o Mang gen deoR đột biến giúp dễ thu nhận mảnh ADN lớn
DH5-ɑ
Vector tạo dòng
Vật liệu di truyền trung gian chuyển và lưu trữ gen tái tổ hợp trong tb chủ
o Có khả năng tự sao chép ở tb chủ o Có thể tồn tại ổn định trong tb chủ qua nhiều thế hệ o Trình tự nhận diện enzym cắt giới hạn đưa gen vào o Mang yếu tố chọn lọc để xác định tb có mang vector o Gồm
• Plasmid
• Vector phage tiêu giải từ phage lamda
• Cosmid
• Vector phage sợi M13
Yêu cầu:
Vector plasmid
o Có yếu tố đánh dấu để chọn lọc dòng tái tổ hợp, thường là
một hay nhiều gen kháng kháng sinh
o Có điểm Ori cho phép nhân bản độc lập NST. Plasmid được hiện nay có điểm Ori có thể kiểm soát được. VD: cho phép khuếch đại plasmid khi có mặt chất ức chế tổng hợp protein (như chloramphenicol) hiệu suất cao khi chiết với độ tinh khiết khá tốt
o Có vùng tạo dòng (MCS): là nhiều trình tự nhận diện duy nhất của các RE khác nhau nằm liên tiếp (polylinker), để cắt mở vòng plasmid và nối đoạn gen mong muốn vào.
Plasmid: ADN kép, vòng, nhân bản độc lập với NST Yêu cầu:
Plasmid được đưa vào tb
chủ = biến nạp, thẩm điện
VD: pUC, pBR322,
YT đánh dấu chèn
pBluescript II
Yếu tố chọn lọc
Các vector plasmid
pUC pBR322 pGEM và pBluescript
pUC
Vector từ phage lamda
Phage tiêu giải
Vector từ phage ʎ
o Kích thước ADN sau tái
o Cắt bỏ một phần bộ gen phage không liên quan đến quá trình tiêu giải và thay bằng đoạn ADN cần tạo dòng tổ hợp: 78% (38 kb) 102% (51 kb) so với dạng hoang dại (50 kb) mới đóng gói được
Tạo vector từ phage:
Vector từ phage lamda
o Đóng gói invitro. Điều này rất có ý nghĩa vì khi đó chỉ cần một lượng ít vector và ADN cần chèn. Đóng gói in vitrolà cách đơn giản nhất để đưa ADN tái tổ hợp vào tế bào E.colichủ.
o Các thư viện phage có thể dễ dàng phát hiện với mẫu
dò, khuếch đại và bảo quản trong một thời gian dài
Ưu điểm:
Vector cosmid
o Mang yếu tố chọn lọc, điểm Ori (~plasmid) o Vị trí tạo dòng, trình tự mã hóa cho vị trí cos(~phage ʎ)
Cosmid = cos site + plasmid: dạng lai phage và plasmid
Vector cosmid
o Cắt vector bằng RE tại MCS và trộn với đoạn gen muốn
chèn (có chiều dài từ 35 - 45 kb)
o Vector và đoạn chèn sẽ được nối lại bằng ligase, tạo sợi ADN thẳng với đoạn chèn bị kẹp giữa 2 phần của vector và tận cùng với các vị trí cosở 2 đầu
o Đóng gói invitronhờ nhận diện 2 vị trí cos:Sợi ADN này ủ với các protein vỏ sẽ tạo thành phage và được gây nhiễm vào E.coli
o Sau khi vào tế bào chủ, ADN tái tổ hợp sẽ tạo thành vòng
nhờ vị trí cosvà hoạt động như một plasmid
Tạo vector cosmid:
Ƣu điểm
Tạo dòng các đoạn gen có kích thước lớn đến 50 kb,
trong khi đó plasmid và phage lambda < 15 kb
Các phƣơng pháp & kỹ thuật cơ bản
•Chiết tách và tinh chế vật liệu di truyền
•Lai acid nucleic
•Kỹ thuật PCR
•Tái tổ hợp và tạo dòng gen
•Xác định trình tự acid nucleic
Chiết tách và tinh chế ADN
Nguyên tắc
Phá vỡ tế bào phóng thích VLDT + tế bào chất Tủa các thành phần trong tbc và để lại ADN tan Cô đặc và thu hồi ADN dạng tinh khiết
Phá thành + màng tb bằng pp vật lý/ hóa học
Phá vỡ tb vật lý/ hóa học
Chiết bằng phenol/chloroform
- Biến tính protein và phân tách cùng pha hữu cơ, để lại ADN tan trong nước - Loại ARN: ribonuclease (RNase)
Thu hồi acid nucleic
- Tủa ADN bằng ethanol tuyệt đối với sự hiện diện muối cation hóa trị I, nhiệt độ thấp - Isopropanol đồng thể tích với pha nước
Chiết ADN bộ gen tế bào
o Tb thực vật: nghiền tb trong cối đá o Tb động vật: enzym, chất tẩy
Phá vỡ tế bào
o Thay phenol/chloroform = CTAB. CTAB tạo phức không
tan với ADN, đường và lipid tan o Tách ADN bằng sắc ký hấp phụ
Tách ADN
Chiết plasmid
o Phá vỡ tb trong điều kiện nhẹ nhàng, ADN NST không đứt đoạn nên kích thước lớn và gắn màng tb nhờ đó loại bỏ bằng ly tâm
Cấu dạng:
o Phá vỡ tb bằng SDS, ADN NST đứt đoạn o Biến tính ADN thẳng từ sợi kép đơn bằng pH 12, ADN
plasmid siêu xoắn không ảnh hưởng
o Thêm acid, ADN NST hồi tính không chính xác nên tủa
Tách plasmid khỏi ADN NST: Kích thước:
Chiết ADN phage
Chiết tách từ thực khuẩn chứ không từ tb chủ nên chiết tách khá đơn giản. Cần loại bỏ capsid của phage o Ly tâm loại tb chủ o Tủa phage bằng PEG o Loại capsid bằng phenol
Tinh chế acid nucleic
Ly tâm phân đoạn: lượng acid nucleic lớn. Ly tâm phân
tách theo thang tỷ trọng saccharose hoặc CsCl2
o Lọc gel: tách ADN qui mô lớn o HPLC: tách oligonucleotid, độ phân giải cao đến 1 nu o Hấp phụ: mARN với đuôi polyA được hấp phụ bằng resin
Điện di: tách ADN qui mô nhỏ
o Gel agarose o Gel polyacrilamide: tinh chế acid nucleic với khác biệt 1 nu
Sắc ký:
Định tính & định lƣợng ADN
Định lƣợng ADN – Quang phổ kế
o 50 μg/ml dung dịch ADN (hoặc ARN) sợi đôi o 40 μg/ml dung dịch ARN (hoặc ADN) sợi đơn o 30 μg/ml oligonucleotid (tới 70 base)
Định lượng: ADN hấp thu cực đại 257nm OD260nm 1 OD260nm tương đương
Kiểm tra độ tinh khiết: tỷ lệ OD260/230 hoặc OD260/280
Định tính ADN – Điện di gel
Phân tách acid nucleic/ protein dưới tác động của điện trường xuyên qua 1 hệ gel. Các phân tử sẽ tách ra: o Tùy theo kích thước o So sánh với chuẩn
Kỹ thuật lai acid nucleic
Khái niệm
Lai phân tử: 2 ptử nucleic sợi đơn trong điều kiện thích hợp có thể bắt cặp bổ sung bằng lk H2 tạo sợi kép
o = lai phân tử o Sản phẩm: phân tử lai
(duplex)
o Phản ứng thuận nghịch o Kép đơn: biến tính
bởi to/ hóa chất
o Lai đặc hiệu: bắt cặp bổ sung hoàn toàn bền
Đặc điểm
Yếu tố ảnh hƣởng
Nhiệt độ: quyết định độ bền lk H2 Độ dài trình tự lai Tỷ lệ GC trong duplex Nồng độ ion Nồng độ phân tử, thời gian phản ứng
Nhiệt độ chảy
Tm: Nhiệt độ tại đó 50% duplex hình thành. Phụ thuộc chiều dài, tỷ lệ GC, nồng độ ion…
To > Tm: biến tính duplex To < Tm: phản ứng lai (hồi tính)
Tm (oC) = 4(GC) + 2(AT)
Kỹ thuật lai
Đoạn dò (probe): oligonucleotid có trình tự bổ sung với phân tử đích, được đánh dấu bằng phóng xạ, quang, huỳnh enzym….
Nguyên tắc: Dùng đoạn dò để lai và phát hiện ra phân tử đích. Nếu có sự tồn tại phân tử lai có phân tử đích trong mẫu
Kỹ thuật lai
Phản ứng lai tiến hành trong điều kiện sao cho sự lai diễn ra đặc hiệu: o Đặc hiệu duplex bền phát hiện o Không đặc hiệu biến tính không phát hiện
Phân loại
Lai sau khi điện di, phát hiện và xác định kích thước phân tử đích
Lai tại chỗ: định vị acid trong mô/tb Lai trong dung dịch, duplex được tách khỏi dung dịch để phát hiện chẩn đoán bệnh dựa trên acid nucleic
Lai trên màng rắn: phân tử tham gia được cố định trên màng nitrocellulose, probe tự do trong dung dịch duplex gắn trên màng o Southern blot: đích là ADN o Northern blot: đích là ARN o Dot blot: hỗn hợp acid nucleic chấm trực tiếp trên màng
l
t o b n r e h t u o S
Southern blot
Southern blot
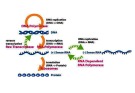
Kỹ thuật PCR
Nguyên lý
o Trong điều kiện có sẵn nucleotid o Enzym polymerase sẽ kéo dài mạch ADN theo nguyên tắc
bổ sung nếu đầu 3’-OH trống
Dựa trên sao chép ADN invitro
Tiến trình PCR
o Biến tính: dsADN ssADN nhờ nhiệt độ o Gắn mồi: ủ các đoạn mồi (chuỗi oligonucleotid ADN tổng hợp đặc hiệu, gồm mồi xuôi, mồi ngược) với các ssADN o Kéo dài: enzym kéo dài mồi theo NTBS với khuôn mẫu
Sự lặp lại của 1 tiến trình gồm 3 bước:
3’
5’
Chƣơng trình PCR
Biến tính khởi đầu: 95oC / 5-10 phút Chu kỳ nhiệt: gồm 3 bước lặp lại 25-40 lần
o Biến tính: 94-95oC / 30s-vài phút o Gắn mồi: 40-70oC (< Tm của mồi) / 30s-60s o Kéo dài: 65-74OC / thời gian tùy theo độ dài khuôn Kéo dài kết thúc: 72-74oC / vài lần thời gian kéo dài
Thành phần phản ứng:
• Khuôn mẫu • Mồi • Đệm: pH thích hợp và nồng độ Mg++ thay đổi • dNTPs: A, T, G và C • DNA polymerase
Yêu cầu thành phần phản ứng
o 18-30 nu, bổ sung đặc hiệu với mạch khuôn o Tỷ lệ GC 40-60% o Không tạo nút kẹp tóc, 2 mồi không bắt cặp với nhau o Tính Tm Nhiệt độ gắn mồi < Tm (oC) 3-5oC
Mồi
o Chịu nhiệt, nguồn gốc từ VSV ưa nhiệt o VD: Taqpolymerase
Polymerase
5’
5’
Ƣu nhƣợc điểm
o Phải biết trình tự 2 đầu để thiết kế mồi o Ngoại nhiễm mất tính đặc hiệu
Ưu: Nhanh, Đơn giản, Nhạy, Đặc hiệu Nhược
o Nested PCR (mồi tổ) o RT-PCR: PCR ngược, dùng ARN là khuôn tổng hợp ADN o Realtime-PCR: phát hiện và định lượng sp bằng huỳnh
quang số lượng ADN khuôn ban đầu
• Nhuộm xen giữa
• Phân hủy đoạn dò
• Lai hóa đoạn dò
Cải tiến
Nhuộm xen giữa
Hủy đoạn dò
Lai hóa đoạn dò
Ứng dụng
Khuếch đại ADN dùng trong nghiên cứu, tạo dòng gen Phát hiện ADN trong chẩn đoán, với độ nhạy và độ
Xác định huyết thống, pháp y
đặc hiệu cao
Tái tổ hợp và tạo dòng gen
Nguyên lý
Khác PCR: tạo dòng gen chưa biết cấu trúc, do đó
Cô lập gen số lượng lớn ở dạng tinh khiết để nghiên cứu cấu trúc, cần đưa vào tb chủ để nhân bản Tạo dòng gen
thực hiện trước khi PCR
o Tạo đoạn ADN o Nối đoạn ADN cần tạo dòng vào vector tạo phân tử tái tổ
hợp
o Đưa ADN tái tổ hợp vào tb chủ o Chọn lọc dòng tái tổ hợp
Gồm
Tạo đoạn gen mong muốn
o Cắt phân tử ADN lớn thành các đoạn nhỏ. Điều này thường được sử
dụng khi xây dựng các thư viện gen
o Sử dụng 2 loại RE khác nhau để các đoạn tạo ra có kích thước vừa phải cho việc tạo dòng phân tích các đoạn thu được và lập bản đồ các vị trí cắt của bộ gen
o Lưu ý vị trí cắt sẽ tạo ra đầu tù/ đầu dính phù hợp với vector mới nối
vào được
Dùng RE
o Nếu cấu trúc đoạn gen đã được biết o Đoạn ADN thu được bằng PCR có các đầu bị so le (lệch) không xác
định về trình tự
o Làm tà đầu sản phẩm PCR, gắn linker hoặc adaptor o Thiết kế mồi có chứa trình tự nhận diện cắt của RE
Kỹ thuật PCR
Nối ADN
Đầu tù:
o Linker (đoạn nối): Oligonucleotid sợi đôi tổng hợp ngắn có
đầu tà và chứa trình tự cắt (đầu dính) của một RE
o Adaptor: khắc phục nhược điểm linker, đây là một
oligonucleotid sợi đôi tổng hợp có sẵn một đầu dính
o Tạo đuôi homopolymer: dùng enzym doxynucleotidyl transferase để thêm một loạt các nucleotid cùng loại vào đầu 3’-OH của sợi ADN tạo thành đuôi homopolymer
Dùng ligase của T4 (tinh chế từ E.colinhiễm T4) Trộn vector duỗi thẳng và ADN với ligase trong điều kiện thích hợp lk phosphodiester và nối các ADN lại
Homopolymer
Linker
Chuyển ADN vào tb chủ
để ADN lọt qua
Biến nạp: Vk bắt ADN từ môi trường Tải nạp: sử dụng thực khuẩn để gây nhiễm tế bào đích Thẩm điện: dùng điện thế cao làm thủng màng tế bào
Chuyển nhiễm: cơ chế giống biến nạp nhưng có thể
Đạn sinh học: “súng bắn gen” đưa ADN gắn trên hạt
áp dụng cho tế bào động vật
vàng xuyên qua màng tế bào
Chọn lọc dòng tái tổ hợp
Kỹ thuật bổ sung anpha Sử dụng sự đề kháng kháng sinh Chọn lọc trực tiếp Lai khóm
Xác định trình tự acid nucleic
Giải trình tự
So sánh
Sanger-Coulson
Maxam-Gilbert
Hiệu quả
Cao
Kém
Độc hại
Ít
Độc
Mồi
Cần
Không
2 phương pháp: Sanger – Coulson Maxam – Gilbert
Phƣơng pháp Sanger-Coulson
PP enzym/dideoxy: dựa vào sự ngừng tổng hợp ADN
khi gặp phải dideoxynucleotid (Frederick Sanger)
o Thực hiện 4 phản ứng PCR riêng biệt với 1 mồi:
A: có dNTP + ddATP
T: có dNTP + ddTTP
G: có dNTP + ddGTP
C: có dNTP + ddCTP
o Mỗi ống sản phẩm được nạp vào một giếng điện di o Kết quả điện di được đọc trình tự
Qui trình:
Đọc kết quả Phƣơng pháp Sanger
5’-GGCCGCGTCTTCGCCGTAG-3’
ddG ddA ddT ddC
3’
5’
Phương pháp Sanger- Coulson cải tiến
Phƣơng pháp Maxam-Gilbert
o Tạo ADN sợi đơn từ đoạn ADN cần giải trình tự o Đánh dấu các phân tử ADN ở một đầu o Thực hiện 4 ống phản ứng riêng biệt:
G: + dimethyl sulphate Guanine bị methyl hóa
AG: + formic acid Adenin và Guanine bị methyl hóa
TC: + Hydrazine biến đổi Thymin và Cytosin
C: + Hydrazine + 1,5 M NaCl biến đổi Cytosin
o Xử lý 4 ống trên với piperidine 1M/90oC để cắt ADN ở liên
kết phosphat kế nucleotid bị biến đổi o Mỗi ống được nạp vào một giếng điện di o Kết quả điện di được đọc và biện luận trình tự
PP hóa học: dựa vào sự thủy giải đặc trưng của ADN












![Tài liệu giảng dạy Sinh học và di truyền [mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260323/hoatudang2026/135x160/42181774414220.jpg)

![Giáo trình Công nghệ vi sinh (Nghề Công nghệ sinh học TC/CĐ) - Trường Cao đẳng Đà Lạt [Mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260224/hoacattuong2026/135x160/87621772161812.jpg)






![Giáo trình Vi sinh vật học môi trường Phần 1: [Thêm thông tin chi tiết nếu có để tối ưu SEO]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20251015/khanhchi0906/135x160/45461768548101.jpg)


