
Characterization of the molten globule state of
retinol-binding protein using a molecular dynamics
simulation approach
Emanuele Paci
1
, Lesley H. Greene
2
, Rachel M. Jones
2
and Lorna J. Smith
2
1 Institute of Molecular Biophysics, School of Physics and Astronomy, University of Leeds, UK
2 Department of Chemistry and Oxford Centre for Molecular Sciences, Chemistry Research Laboratory, University of Oxford, UK
The detailed characterization of molten globule states
of proteins continues to be an area of intense research
activity and interest (for reviews see [1,2]). This is in
part because these equilibrium partially folded states
have been found to have many similarities to kinetic
protein folding intermediates. As such, the properties
of these states can therefore give important insights
into the determinants of protein structure and folding
[2]. Molten globule states of proteins are also postula-
ted to be involved in a range of important physiologi-
cal processes, including the insertion of proteins into
membranes, the release of bound ligands and aggrega-
tion [1]. In this latter area, molten globule-like species
are thought in some systems to be precursors of amy-
loid fibril formation [3,4].
Molten globule ensembles are characterized by hav-
ing a pronounced amount of secondary structure, in a
compact state that lacks most of the specific tertiary
interactions coming from tightly packed side chain
groups [1,2]. One of the molten globule states that has
been studied in the most depth is that of a-lactalbumin
[5]. In this case, it has been possible using nuclear
magnetic resonance (NMR) methods to gain a residue
specific picture of the noncooperative unfolding of the
molten globule during denaturation with urea [6–8];
data from these experiments have also been used as
Keywords
lipocalin; molecular dynamics; molten
globule; protein folding; retinol-binding
protein
Correspondence
L. J. Smith, Department of Chemistry,
University of Oxford, Chemistry Research
Laboratory, Mansfield Road, Oxford OX1
3TA, UK
Fax: +44 1865 285002
Tel: +44 1865 275961
E-mail: lorna.smith@chem.ox.ac.uk
E. Paci, Institute of Molecular Biophysics,
School of Physics and Astronomy,
University of Leeds, Leeds LS2 9JT, UK
Tel: +44 113 3433806
E-mail: e.paci@leeds.ac.uk
(Received 25 May 2005, revised 12 July
2005, accepted 3 August 2005)
doi:10.1111/j.1742-4658.2005.04898.x
Retinol-binding protein transports retinol, and circulates in the plasma as a
macromolecular complex with the protein transthyretin. Under acidic con-
ditions retinol-binding protein undergoes a transition to the molten globule
state, and releases the bound retinol ligand. A biased molecular dynamics
simulation method has been used to generate models for the ensemble of
conformers populated within this molten globule state. Simulation con-
formers, with a radius of gyration at least 1.1 A
˚greater than that of the
native state, contain on average 37% b-sheet secondary structure. In these
conformers the central regions of the two orthogonal b-sheets that make
up the b-barrel in the native protein are highly persistent. However, there
are sizable fluctuations for residues in the outer regions of the b-sheets,
and large variations in side chain packing even in the protein core. Signifi-
cant conformational changes are seen in the simulation conformers for resi-
dues 85–104 (b-strands E and F and the E-F loop). These changes give an
opening of the retinol-binding site. Comparisons with experimental data
suggest that the unfolding in this region may provide a mechanism by
which the complex of retinol-binding protein and transthyretin dissociates,
and retinol is released at the cell surface.
Abbreviations
ANS, 8-anilino-1-napthalenesulphonate; MD, molecular dynamics; RBP, retinol-binding protein; R
g
, radius of gyration; RMSD, root-mean-
square deviation; TTR, transthyretin.
4826 FEBS Journal 272 (2005) 4826–4838 ª2005 FEBS

restraints in a novel approach to determine the free
energy landscape of this molten globule [9]. In the
majority of the proteins whose molten globule states
have been characterized, both by experimental and the-
oretical methods, predominantly a-helical secondary
structure persists in the molten globule state [10–13].
In contrast, in this paper we concentrate on a protein
which is rich in b-sheet secondary structure in the
native state, and which retains the majority of this
b-sheet secondary structure in the molten globule state,
human serum retinol-binding protein (RBP).
RBP is a member of the lipocalin superfamily [14].
The proteins in this superfamily adopt a similar fold;
an eight-stranded up-and-down b-barrel with a C-ter-
minal helix. However, they have a wide range of func-
tions, and high levels of sequence divergence, with
many members sharing under 20% sequence identity
[14,15]. The lipocalins are widely distributed through-
out the eukaryotic and prokaryotic kingdoms [16,17].
Many of the lipocalins act as transporters for small
nonpolar ligands, such as retinoids, haem, phero-
mones, lipids, prostaglandins and pigments [18]. RBP
transports all-trans-retinol (vitamin A) from its storage
sites in the liver to target tissues [19]. It is postulated
that the local decrease in pH at the surface membrane
of target cells triggers the release of retinol, by a mech-
anism that is dependent on the conversion of RBP to
a molten globule state [20,21]. It is possible that similar
mechanisms may prompt ligand release for other mem-
bers of the lipocalin superfamily. For example, the
release of lipid ligands by human tear lipocalins under
acidic conditions is thought to be associated with a
transition to the molten globule state [22,23].
The molten globule state of RBP, formed under acidic
conditions, has been shown to exhibit the key character-
istics typical of these partially folded states. Stoke’s
radii, from diffusion coefficient measurements, have
demonstrated that the molten globule retains a compact
fold [20]. The mean molecular dimensions of the parti-
ally folded ensemble are only 13% larger than those
of the native state. Far- and near-UV circular dichroism
(CD) spectra show that the protein contains a significant
level of secondary structure, but has a considerable level
of disorder in side chain packing, respectively [20,24,25].
Obtaining detailed information at an atomic level about
the molten globule state using techniques such as NMR
spectroscopy is challenging. Partially folded states such
as molten globules are ensemble of interconverting con-
formers [26]. Slow interconversion between populated
conformers gives rise to broadened NMR resonances,
while averaging of chemical shifts across the populated
ensemble gives a limited chemical shift dispersion [6,27].
Therefore, to develop a detailed model to further our
understanding of the molten globule state of RBP,
in vitro and in vivo experimental studies have been com-
plemented with a molecular dynamics (MD) simulation
study. The results of this are reported here.
It is not possible to explore adequately the conform-
ational space accessible to partially folded proteins,
within the simulation timescale currently accessible to
conventional MD simulations of proteins in explicit
solvent. The exploration of non-native conformations
is therefore usually achieved, either by using very high
temperatures in the simulations, or by introducing a
suitable perturbation in a biased MD simulation, often
using implicit solvent models (for a review of this topic
see, e.g [28,29]). Explicitly modelling the perturbation
induced by a change in the solution pH would not
prompt the transition from the native to the molten
globule state, on a timescale which can be directly
simulated. In this work therefore we have used three
different perturbations in turn. One perturbation forces
an increase in the protein radius of gyration, the sec-
ond perturbation induces the breaking of native con-
tacts in the structure and the third perturbation is
aimed to speed up the exploration of diverse (in terms
of mutual RMSD) conformations. These various per-
turbations are applied using a particularly ‘soft’ time-
dependent bias [30,31], designed to generate low energy
pathways in the conformational space. The large num-
ber of diverse and moderately non-native conforma-
tions generated with this biased molecular dynamics
approach are then used as initial conformations for
unperturbed, room temperature simulations. This
method allowed us to explore local free energy minima
in a broad region of the conformational space close to
the native state. The approach is designed to provide a
qualitative map of the free energy landscape, in a
region of the conformation space compatible with the
experimental knowledge of the molten globule state.
By applying this sampling approach to RBP, we are
able to identify a broad basin of low energy partially
folded conformers that are compatible with the avail-
able experimental data [20,24,25,32–34]. These con-
formers provide a model for the molten globule state
of RBP that allows us to gain insight into the determi-
nants of protein folding and the mechanism of retinol
delivery and release, an important physiological prob-
lem which remains unresolved.
Results and Discussion
Sampling of conformational space
As described in the Experimental procedures section,
a biased MD simulation method has been used to
E. Paci et al. MD simulations of the RBP molten globule state
FEBS Journal 272 (2005) 4826–4838 ª2005 FEBS 4827
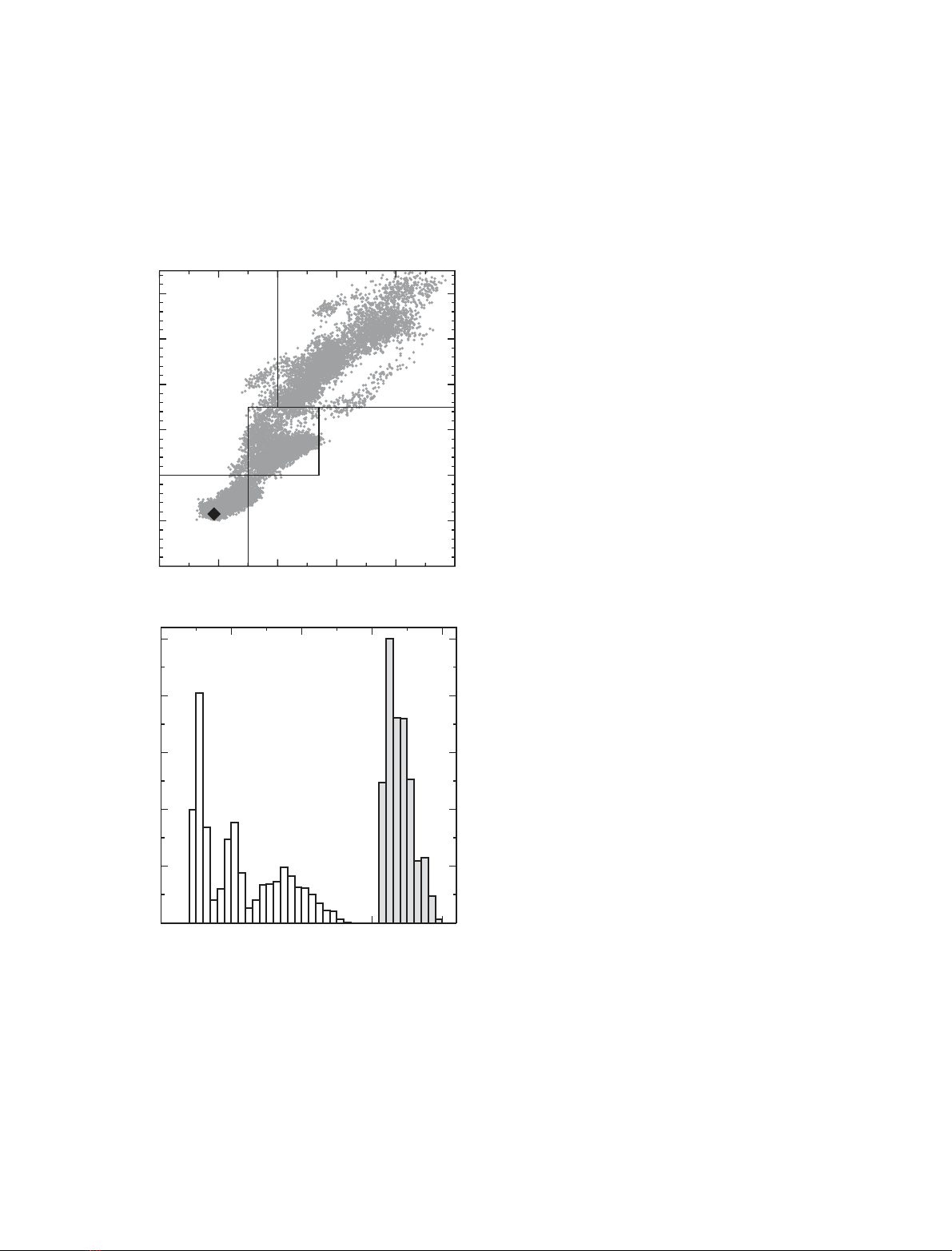
generate 160 diverse configurations of RBP. Each of
these was then used as an initial configuration in an
unbiased MD simulation of 1.2-ns length. Figure 1(A)
shows a plot of the heavy atom RMSD (root-mean-
square deviation) from the X-ray structure as a func-
tion of the radius of gyration (R
g
), for structures taken
every 10 ps through the 160 unbiased MD simulations.
These data demonstrate the broadness of the conform-
ational space explored in the study. Analysis of the
range of RMSD values seen for the MD conformers
shows that there are three distinct peaks in the RMSD
distribution (Fig. 1B). These correspond to different
levels of unfolding. To aid the analysis, the conformers
have been divided into three groups on the basis of
their RMSD values. Conformers in group 1 have an
RMSD less than 4 A
˚and an R
g
less than 16.5 A
˚.
Group 2 conformers have an RMSD in the range
4–7 A
˚and an R
g
in the range 16.5–17.7 A
˚, while
group 3 conformers have an RMSD above 7 A
˚and an
R
g
greater than 17 A
˚. The characteristics of the group
1 conformers are very native-like, in keeping with their
low RMSD values (< 4 A
˚). For example, 81 of the
residues that are in regions of secondary structure in
the native protein have secondary structure popula-
tions greater than 0.8 in the group 1 ensemble of con-
formers. We therefore focus our attention on the
conformers in groups 2 and 3 that display a greater
level of unfolding. The native state structure of RBP
has an R
g
of 15.9 A
˚. The 13% increase in molecular
dimensions seen experimentally on forming the molten
globule state would correspond to an effective R
g
of
18 A
˚for the molten globule ensemble. The group 3
conformers, with an R
g
in the range 17–20 A
˚, show an
appropriate level of expansion on average for the mol-
ten globule state (Fig. 1A). The properties of this
group of conformers have therefore particularly been
compared with experimental data for this state.
Secondary structure persistence
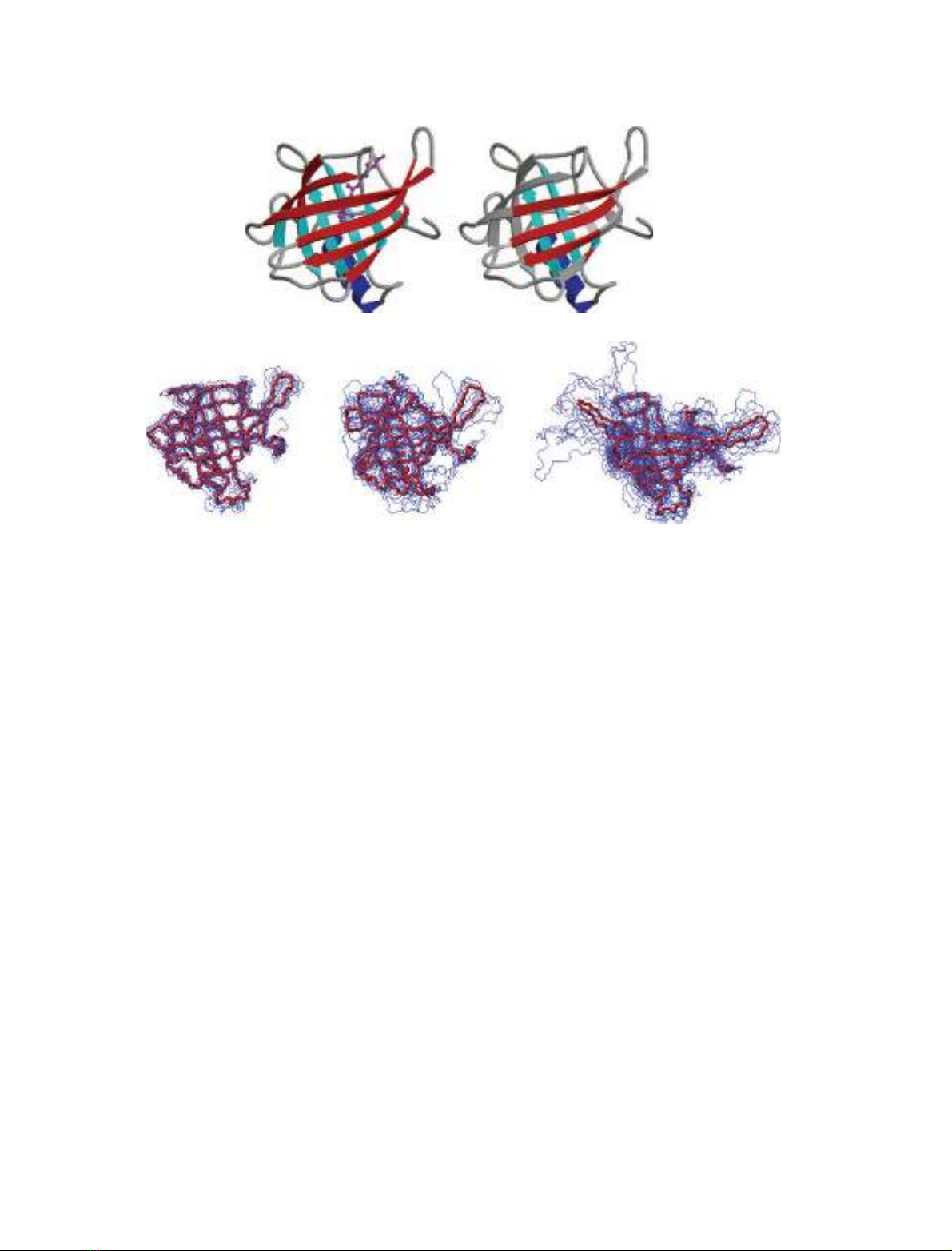
The b-barrel in the native structure of RBP consists of
eight b-strands (A-H) [35]. These are arranged in two
orthogonal b-sheets, with some of the b-strands being
involved in both of the sheets. The first sheet consists
of strands ABCDEF, and the second sheet contains
strands EFGHA (Fig. 2). The native structure also
contains an a-helix (residues 146–158), which packs
onto the b-sheet formed by the second set of strands.
All of the b-strands present in native RBP show a high
level of persistence in the group 2 conformers (Figs 2
and 3). However, in many of the structures some of
these strands are reduced in length, or have irregularit-
ies compared to those in the native state. For example,
for strand F (residues 100–109) the mean b-sheet
populations for the first five residues are only 0.05–
0.15. For strand E (residues 85–92) the mean b-sheet
populations for the terminal residues 85 and 91–92 are
0.05–0.13, while those for residues 86–90 are 0.71–0.84.
The disruption of this b-strand results predominantly
0510 15 20
Å
0
0.1
0.2
0.3
0.4
0.5
normalized probability
RMSD
Rg
B
15 16 17 18 19 20
Rg (Å)
0
2
4
6
8
10
12
RMSD (Å)
Group2
Group3
Group1
A
Fig. 1. (A) Relationship between the protein radius of gyration and
the RMSD value from the X-ray structure of native RBP, for the
16 000 conformers taken at 10-ps intervals along the unbiased
simulations. The radius of gyration and RMSD values are calculated
using all heavy atoms. The black diamond corresponds to the native
state following the 2 ns equilibration simulation. The definitions of
the three groups of conformers used in the analysis are shown. (B)
The distribution of radius of gyration (filled bars) and RMSD values
(open bars) across the 16 000 simulation conformers.
MD simulations of the RBP molten globule state E. Paci et al.
4828 FEBS Journal 272 (2005) 4826–4838 ª2005 FEBS
from the loss of hydrogen bonds between strands E
and F, although in some structures the hydrogen
bonds between strand D and E are also missing. The
a-helix also shows a high level of persistence in the
group 2 conformers, with a-helical populations in
the range 0.59–0.99 for the residues involved.
In the group 3 conformers the changes are more
pronounced (Figs 2 and 3, and Fig. 1 in the supple-
mentary material). All the b-strands now show reduc-
tions in length compared to those in the native
structure. Even strand H (residues 129–138), one of
the particularly persistent strands, is reduced in length
by three residues in more than half of the structures,
with residues 129 and 130 having b-sheet populations
of less than 0.1. In addition, in group 3 strand E and
the first half of strand F are almost completely lost.
Residues 85–92 and 100–104 show b-sheet populations
of less than 0.3. A significant disorder is seen across
the ensemble of group 3 conformers in the region con-
taining strand E, the first part of strand F and the con-
necting E–F loop (residues 85–104). The changes in
this region correlate with results from crystallographic
studies of bovine RBP. These report a conformational
change in the E–F loop at low pH [34]. In addition,
changes in this region were reported in a simulation of
the apo form of RBP reported previously [36].
In almost all the group 3 conformers, however, a
central region of the b-sheets is preserved. Residues in
the central regions of strands B, C, D, G and H and
part of strand A have b-strand populations greater
than 0.9. A persistent section comprising the central
regions of strands B, C and D together with part of
strand A in b-sheet 1, and the C-terminal region of
strand F with the central parts of strands G and H in
b-sheet 2, have residues with b-strand populations
greater than 0.8 (Figs 2 and 3). Hence in the group 3
conformers the central area of each of the two ortho-
gonal b-sheets, that make up the b-barrel in the native
protein, are retained. This is interesting as the two
parts of the polypeptide chain that form these persist-
ent central regions of the b-sheets have closely similar
amino acid sequences. In particular, the sequence of
RBP contains an internal repeat with residues 36–83
(includes b-strands BCD) and 96–141 (includes
b-strands FGH) having 34% identity [35]. This may
account, at least in part, for the similar behaviour of
these regions in the simulations. The central region
of the a-helix is also very persistent in the group 3
A
B
C
D
E
FG
H
AB
C
Fig. 2. (A and B) The X-ray structure of human serum retinol-binding protein [35]. (A) The b-strands are labelled, those in b-sheet 1 are
shown in red and those in b-sheet 2 are shown in cyan. The a-helix is blue and the retinol is magenta. (B) Only the residues that have a sec-
ondary structure persistence greater than 0.8 in the group 3 conformers (Fig. 3) are coloured. The figure was generated using the program
MOLSCRIPT [53]. (C) Backbone trace of representative structures from the three groups of simulation conformers (left, group 1; centre, group
2; right, group 3). In each case the average structure over the cluster centres is shown in red.
E. Paci et al. MD simulations of the RBP molten globule state
FEBS Journal 272 (2005) 4826–4838 ª2005 FEBS 4829
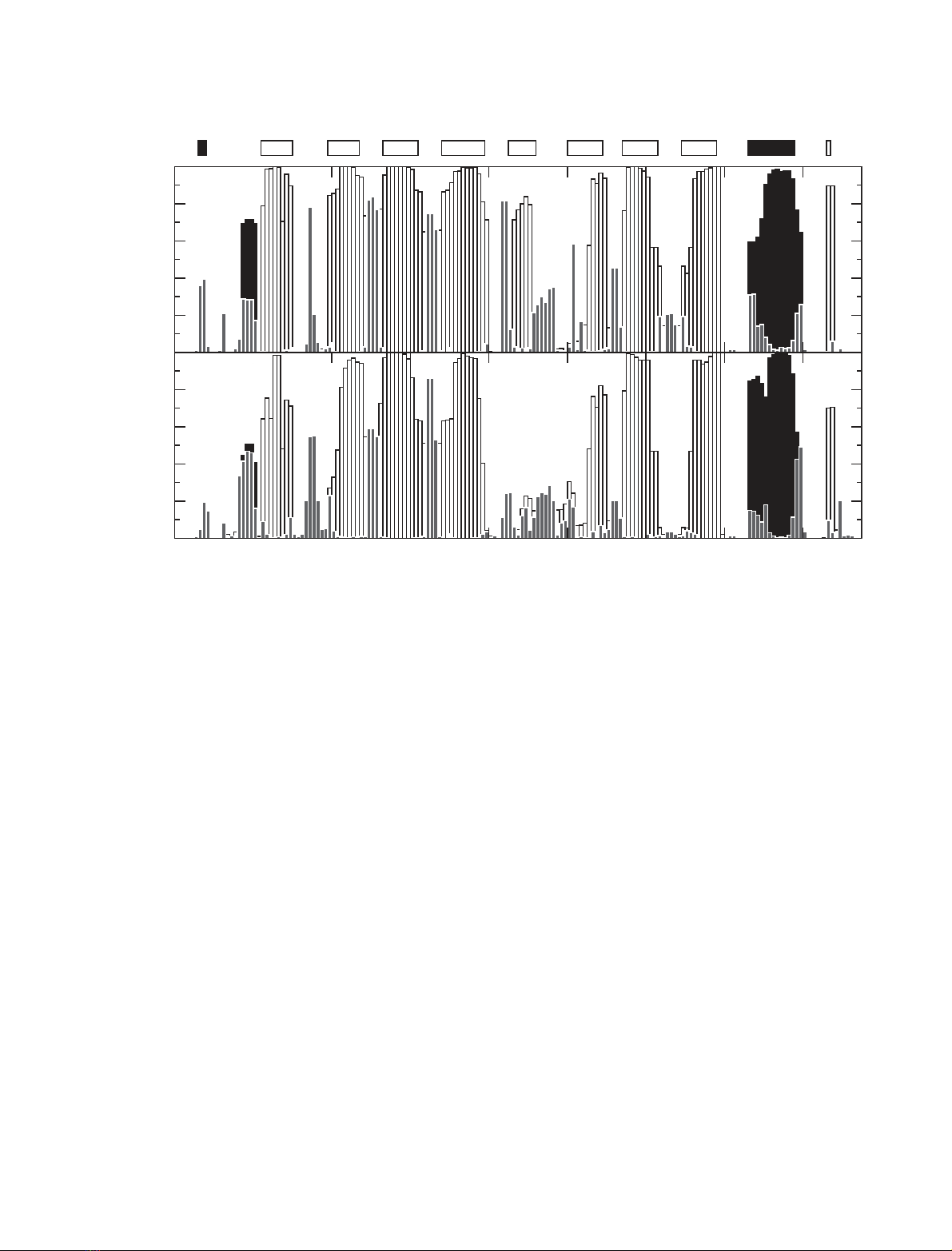
conformers, residues 151–156 having a-helical popula-
tions greater than 0.9.
Experimental estimates of secondary structure con-
tent from far-UV CD spectra give 45 and 40% b-sheet
for the native and molten globule states of RBP,
respectively [26]. The b-sheet estimate for the native-
state is in close accord with that observed in the X-ray
structure (46%) [35]. In the group 3 simulation con-
formers, 53 residues have a b-sheet population of 0.60
or greater, and 11 residues have a b-sheet population
in the range 0.40–0.60. Taken together this corres-
ponds to 37% of residues in b-strand secondary struc-
ture, a value similar to that seen experimentally for the
molten globule state. The experimental CD data show
that there is an increase in a-helical secondary struc-
ture on forming the molten globule state (8% native;
24% molten globule [25]). A large increase in a-helical
secondary structure is not observed, on average, in the
simulation conformers. This difference may reflect
sampling and force field limitations in the simulations,
and the difficulty of interpreting experimental CD data
in a quantitative fashion. The difference may also
reflect the fact that we do not model explicitly the con-
ditions under which the molten globule is stable in the
simulations, but rather identify conformers that are
low in energy under native conditions. However,
although there is not a large increase in a-helical sec-
ondary structure, the native state a-helix for residues
146–158 is essentially retained in all the simulation
conformers. In addition, in some of the conformers,
particularly those in group 2, turns, some of a helical
character, do form for residues 93–96. These residues
are in the loop connecting strands E and F in the
native protein, a region where the native structure is
significantly disrupted in the simulations. It is therefore
possible that this is the part of the RBP sequence that
forms non-native helical secondary structure when the
molten globule state is adopted.
Variations in side chain packing
Despite the high persistence of the central regions of
b-sheet secondary structure even in the conformers
in group 3, significant changes are observed in the
0
0.2
0.4
0.6
0.8
probability
02040
60 80 100 120 140 160
residue number
0
0.2
0.4
0.6
0.8
ABC D E FGH
Fig. 3. Fraction of the simulation conformers belonging to groups 2 (upper panel) and 3 (lower panel) in which certain secondary structure
elements are present. Secondary structure was calculated using the program DSSPcont [54] which identifies regions of secondary structure
through an analysis of hydrogen bonding patterns. b-sheet secondary structure is shown with open bars, helical (a,3
10
and p) secondary
structure is shown with filled black bars, and turns and bends are shown with grey bars. The secondary structure present in the native state
of RBP is indicated at the top of the figure, with the b-strands labelled A–H (open bars, b-strands; filled bars, a-helices).
MD simulations of the RBP molten globule state E. Paci et al.
4830 FEBS Journal 272 (2005) 4826–4838 ª2005 FEBS

























