
The crystal structure of pyruvate decarboxylase from
Kluyveromyces lactis
Implications for the substrate activation mechanism of this enzyme
Steffen Kutter
1
, Georg Wille
1,
*, Sandy Relle
1
, Manfred S. Weiss
2
, Gerhard Hu
¨bner
1
and
Stephan Ko
¨nig
1
1 Institute for Biochemistry, Department of Biochemistry & Biotechnology, Martin-Luther-University Halle-Wittenberg, Halle (Saale),
Germany
2 European Molecular Biology Laboratory Outstation, Hamburg, Germany
Pyruvate decarboxylase (PDC; EC 4.1.1.1) is a key
enzyme of carbon metabolism at the branching point
between aerobic respiration and anaerobic alcoholic
fermentation. It catalyzes the decarboxylation of pyru-
vate in plants, yeasts and some bacteria by using thi-
amine diphosphate (ThDP) and Mg
2+
as cofactors.
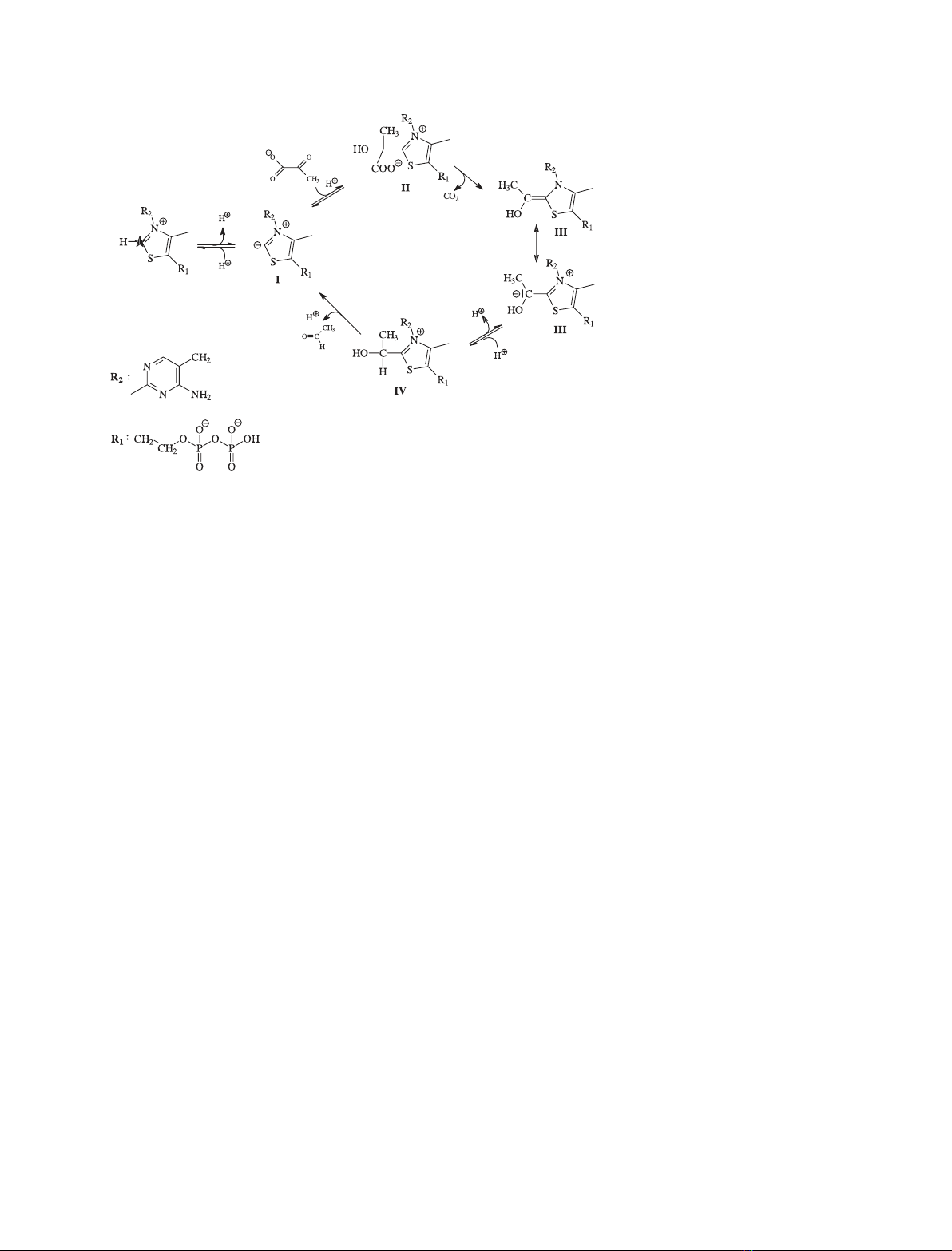
The catalytic cycle of ThDP enzymes is well estab-
lished [1] (Scheme 1). At first, the a-carbonyl group of
the substrate is attacked by the deprotonated C2 atom
of the thiazolium ring of ThDP [the ylid (I)]. In the
case of pyruvate, the resulting lactyl-ThDP (II) is sub-
sequently decarboxylated to yield the central interme-
diate of ThDP catalysis, the a-carbanion ⁄enamine
(III). Protonation of III yields hydroxyethyl-ThDP
(IV), and the release of the second product acetalde-
hyde completes the catalytic cycle of ThDP.
The yeast Kluyveromyces lactis (formerly termed
Saccharomyces lactis) is able to assimilate lactose and
Keywords
allosteric enzyme activation; conformation
equilibrium; disordered loop regions;
thiamine diphosphate
Correspondence
S. Ko
¨nig, Institute for Biochemistry,
Department of Biochemistry &
Biotechnology, Martin-Luther-University
Halle-Wittenberg, Kurt-Mothes-Str. 3,
06120 Halle (Saale), Germany
Fax: +49 345 5527014
Tel: +49 345 5524829
E-mail: koenig@biochemtech.uni-halle.de
*Present address
Institute for Biophysics, Department of
Physics, Johann-Wolfgang-Goethe-University
Frankfurt ⁄Main, Max-von-Laue-Str. 1,
60438 Frankfurt ⁄Main, Germany
(Received 19 June 2006, accepted 13 July
2006)
doi:10.1111/j.1742-4658.2006.05415.x
The crystal structure of pyruvate decarboxylase from Kluyveromyces lactis
has been determined to 2.26 A
˚resolution. Like other yeast enzymes,
Kluyveromyces lactis pyruvate decarboxylase is subject to allosteric sub-
strate activation. Binding of substrate at a regulatory site induces catalytic
activity. This process is accompanied by conformational changes and
subunit rearrangements. In the nonactivated form of the corresponding
enzyme from Saccharomyces cerevisiae, all active sites are solvent accessible
due to the high flexibility of loop regions 106–113 and 292–301. The bind-
ing of the activator pyruvamide arrests these loops. Consequently, two of
four active sites become closed. In Kluyveromyces lactis pyruvate decarb-
oxylase, this half-side closed tetramer is present even without any activator.
However, one of the loops (residues 105–113), which are flexible in nonacti-
vated Saccharomyces cerevisiae pyruvate decarboxylase, remains flexible.
Even though the tetramer assemblies of both enzyme species are different
in the absence of activating agents, their substrate activation kinetics are
similar. This implies an equilibrium between the open and the half-side
closed state of yeast pyruvate decarboxylase tetramers. The completely
open enzyme state is favoured for Saccharomyces cerevisiae pyruvate de-
carboxylase, whereas the half-side closed form is predominant for Kluyve-
romyces lactis pyruvate decarboxylase. Consequently, the structuring of the
flexible loop region 105–113 seems to be the crucial step during the sub-
strate activation process of Kluyveromyces lactis pyruvate decarboxylase.
Abbreviations
KlPDC, pyruvate decarboxylase from Kluyveromyces lactis; PDC, pyruvate decarboxylase; ScPDC, pyruvate decarboxylase from
Saccharomyces cerevisiae; ThDP, thiamine diphosphate.
FEBS Journal 273 (2006) 4199–4209 ª2006 The Authors Journal compilation ª2006 FEBS 4199
convert it to lactic acid. It is commercially utilized for
the production of recombinant chymosin, a proteolytic
enzyme used to coagulate milk in cheese manufac-
turing.
In contrast to S. cerevisiae, only one gene codes for
PDC in Kluyveromyces lactis. The protein (SwissProt
entry Q12629) has 86.3% identical residues and 96.4%
similar residues compared to SwissProt entry P06169,
the dominant PDC in S. cerevisiae [2]. It is known
from small-angle X-ray solution scattering experiments
(unpublished results) that the catalytically active form
of K. lactis PDC (KlPDC) is a homotetramer at micro-
molar protein concentrations (563 amino acid residues
per subunit, total molecular mass 240 kDa). The cofac-
tors ThDP and Mg
2+
are bound tightly, but not cova-
lently, at the interface of two monomers (Fig. 1). At
pH values > 8, the cofactors dissociate from the pro-
tein, resulting in complete loss of catalytic activity.
Lowering the pH to 5.7–6.3, which is also the opti-
mum for KlPDC catalysis, can restore this activity
almost completely.
In 1967, Davies [3] was the first to describe a sigmoi-
dal deviation of the plot of reaction rate vs. substrate
concentration for PDC from wheat germ. Hu
¨bner
et al. [4] established a first model for this substrate
activation phenomenon. Stopped-flow kinetic tech-
niques were used to analyze the substrate activation of
S.cerevisiae PDC (ScPDC). From studies with the
inhibitor glyoxylic acid and the inconvertible activator
pyruvamide (2-oxopropane amide, the amide analog of
the substrate pyruvate), it was concluded that a separ-
ate binding site for the regulatory substrate molecule
must exist. Later, Hu
¨bner and Schellenberger [5]
showed that the enzyme is potentially inactive in the
absence of substrate. With the single exception of the
bacterial enzyme from Zymomonas mobilis [6], all
PDCs studied so far are subject to substrate activa-
tion.
Lu et al. [7,8] described the structural consequences
of substrate activation on the basis of the crystal struc-
ture of pyruvamide-activated ScPDC compared to that
of ScPDC crystallized in the absence of any effectors
[9], which is assumed to be the nonactivated state of
the enzyme. Activation involves a rearrangement of
the two dimers within the tetramer: the D
2
symmetry
of the nonactivated ScPDC is broken, and an open
and a closed side of the tetrameric molecule is formed.
Two different binding sites of the activator were
located: one at the interface between the two domains
within one subunit, and one directly at the active site.
In the presence of pyruvamide, the loop regions 106–
113 and 292–301 undergo a disorder–order transition
and close over the active sites, thus possibly stabilizing
the binding of substrate.
An alternative pathway for substrate activation is
favored by Baburina et al. [10–12] and Li et al. [13,14],
who suggest that an activator molecule, bound to resi-
due Cys221, is the starting point for the activation
transition. However, no electron density for a bound
activator molecule could be detected directly at this
amino acid residue in pyruvamide-activated ScPDC.
Instead, pyruvamide was found to bind 10 A
˚away
from Cys221, in a pocket formed by two of three
domains of the subunit [8].
Scheme 1. Catalytic cycle of pyruvate
decarboxylase. A prerequisite for substrate
binding at the cofactor thiamine diphosphate
(ThDP) is the deprotonation of the C2 atom
of the thiazolium ring (marked by an
asterisk). The resulting ylid of ThDP (I) can
attack the carbon atom of the carbonyl
group of the substrate pyruvate, generating
lactyl ThDP (II), the first tetrahedral
intermediate of the cycle. The subsequent
decarboxylation of II results in the central
reaction intermediate, the a-carbanion-
enamine of ThDP (III). Protonation of III
yields the second tetrahedral intermediate,
the hydroxyl ethyl ThDP (IV). Release of the
second product, acetaldehyde, completes
the cycle.
Crystal structure of pyruvate decarboxylase S. Kutter et al.
4200 FEBS Journal 273 (2006) 4199–4209 ª2006 The Authors Journal compilation ª2006 FEBS
Here, we describe the crystal structure of PDC from
the yeast K. lactis and the structural consequences of
the substrate activation of this PDC species. Our
model constitutes an extension to the activation model
previously proposed and established for ScPDC [8].
Results
Quality of the crystal structure model
The asymmetric unit contains a complete tetramer.
Hence, the final model consists of four polypeptide
chains arranged as a homotetramer of approximate D
2
symmetry. Each monomer was modeled using the
amino acid sequence deduced from KlPDC gene pdc1
[15], corresponding to SwissProt entry Q12629. The
refined model comprises residues 2–105, 114–289 and
303–562 of subunit A, residues 2–104 and 114–554 of
subunit B, residues 2–104 and 116–556 of subunit C,
residues 2–104 and 121–562 of subunit D, four mole-
cules of ThDP, four Mg
2+
, and 1649 water molecules.
The final R-factor is 0.158 (for complete data collec-
tion and processing statistics, see Table 1).
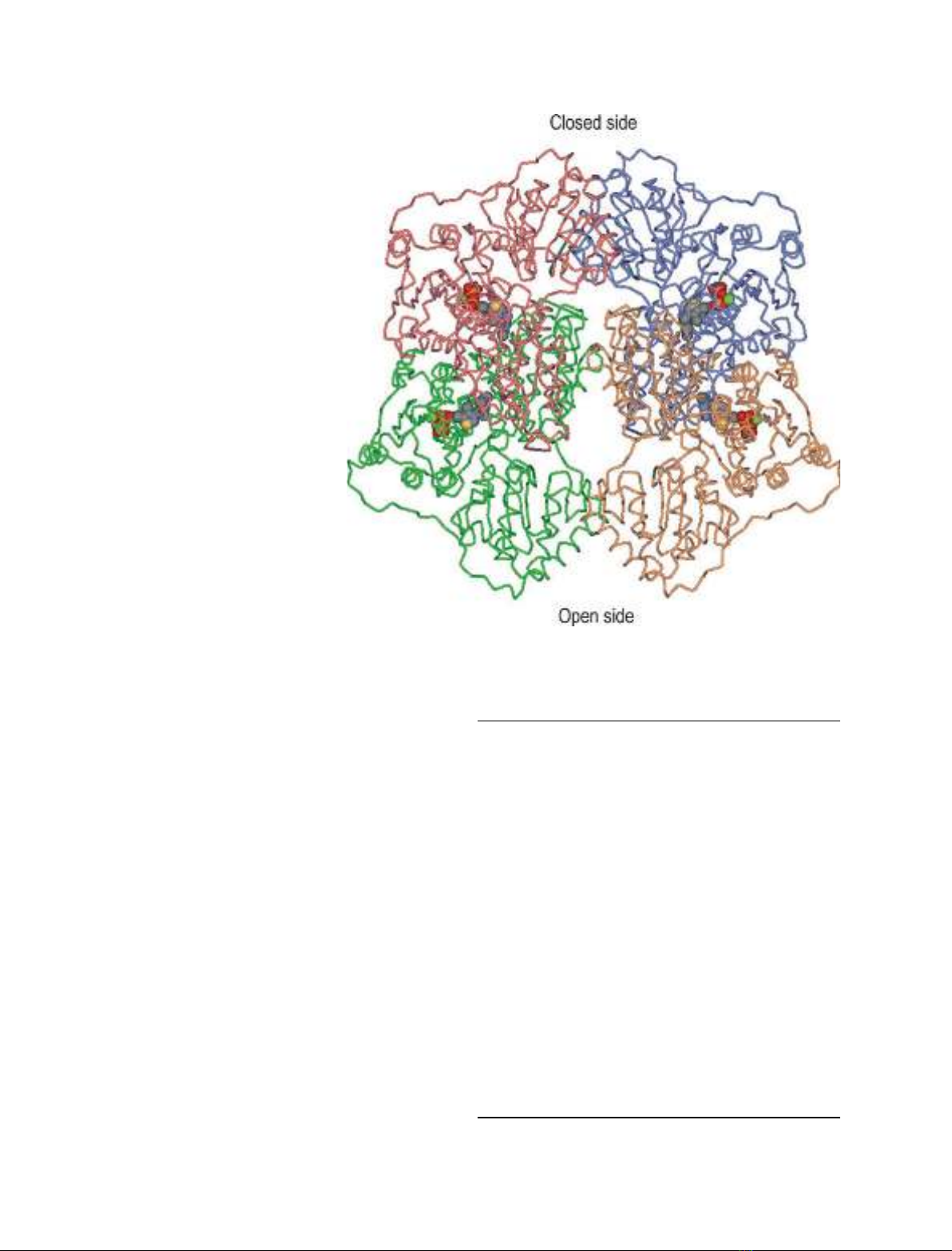
Fig. 1. Catrace of the crystal structure
model of the Kluyveromyces lactis pyruvate
decarboxylase (KlPDC) tetramer. The four
subunits are colored individually (subunit A,
pink; subunit B, green; subunit C, blue;
subunit D, orange). The cofactors thiamine
diphosphate and Mg
2+
(presented in space-
filling mode, colored by their elements,
Mg
2+
in green) are located at the subunit
interface areas (A–B and C–D, respectively)
of both dimers. The open and the closed
side of the tetramer resulting from the spe-
cial dimer arrangement are indicated.
Table 1. Data collection and processing statistics. Values in paren-
theses correspond to the highest-resolution shell.
Number of crystals 1
Beamline X11
Detector MARCCD
Wavelength (A
˚) 0.8125
Temperature (K) 100
Crystal–detector distance (mm) 180
Rotation range per image () 0.5
Total rotation range () 265.5
Space group P2
1
Unit cell parameters (A
˚)a¼78.72, b¼203.09,
c¼79.78, b¼91.82
Mosaicity () 0.40
Resolution limits (A
˚) 99.0–2.26 (2.32–2.26)
Total number of reflections 549 432
Unique reflections 114 899
Redundancy 4.8
I⁄r(I) 20.2 (6.4)
Completeness (%) 98.5 (95.5)
R
merge
(%) 7.1 (21.5)
R
r.i.m.
(%) 8.0 (24.7)
R
p.i.m.
(%) 3.5 (11.8)
Overall B-factor from Wilson plot (A
˚
2
) 28.3
Optical resolution (A
˚) 1.70
S. Kutter et al. Crystal structure of pyruvate decarboxylase
FEBS Journal 273 (2006) 4199–4209 ª2006 The Authors Journal compilation ª2006 FEBS 4201
Neither the terminal residues, nor residues 105–113
in all subunits and residues 290–302 in one subunit,
could be traced in the electron density map, prob-
ably because of too high flexibility of these regions.
Even in subunits B–D, in which the latter region
could be traced, the high flexibility of the loop is
evidenced by B-factors > 50 A
˚
2
, which are clearly
above the average of 22 A
˚
2
(Table 2). In the crys-
tal structure of nonactivated ScPDC, none of the
two loop regions are resolved [9]. However, they are
well defined in the structure of pyruvamide-activated
ScPDC [8]. Another flexible loop in KlPDC is the
one comprising amino acid residues 344–360. This
loop is located at the solvent-exposed surface of the
tetramer and it connects the middle and the C-ter-
minal domains (Fig. 2). In the crystal structure of
pyruvamide-activated ScPDC, the cleft between these
Table 2. Refinement statistics.
Resolution range (A
˚) 23.58–2.26 (2.32–2.26)
Total number of atoms
(nonhydrogen)
18 466
Number of protein atoms 16 776
R
cryst
(%) 15.8 (16.3)
R
free
(%) 21.4 (27.0)
r.m.s.d. from ideality
Bonds (A
˚) 0.015
Angles () 1.477
Ramachandran plot
% in most favored regions 92.5
Average B-factor (A
˚
2
)
Main chain 21.7
Side chain 22.9
Thiamine diphosphate 13.4
Mg
2+
15.8
Water molecules 29.7
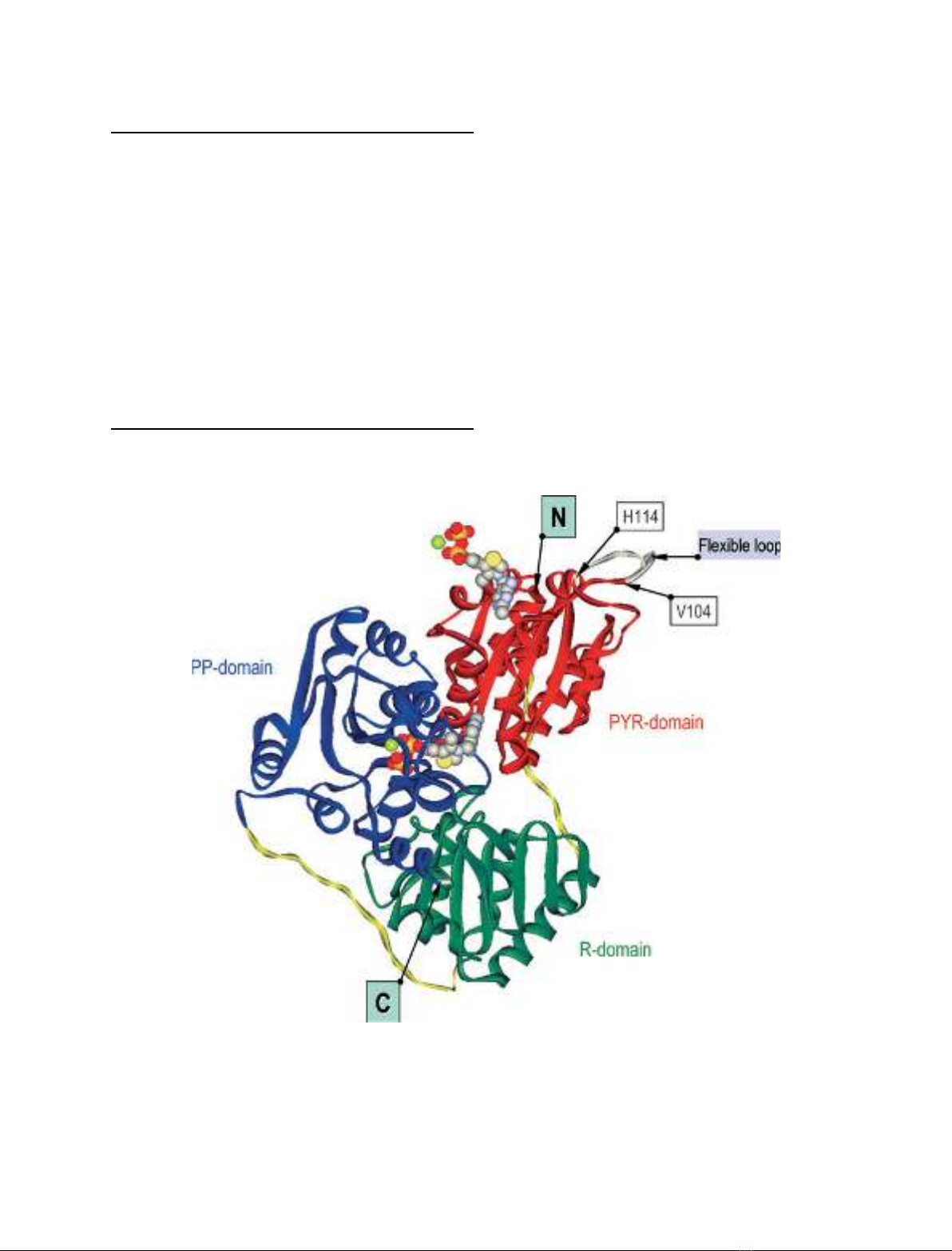
Fig. 2. Ribbon representation of the Kluyveromyces lactis pyruvate decarboxylase (KlPDC) monomer. The domains are colored individually
(N-terminal PYR domain, red; middle R domain, green; C-terminal PP domain, blue; domain-connecting loops, yellow). The cofactors are
depicted in space-filling mode. The positions of the N-terminal and C-terminal amino acid residues of the model, the position of the flexible
loop region, which is omitted in the final model, and the position of the residues adjacent to the loop are labeled. The orientation of the sub-
unit is the same as that of subunit B in Fig. 1.
Crystal structure of pyruvate decarboxylase S. Kutter et al.
4202 FEBS Journal 273 (2006) 4199–4209 ª2006 The Authors Journal compilation ª2006 FEBS
domains contains the binding site for the activator
molecule.
Overall structure
The KlPDC tetramer consists of two asymmetrically
associated identical homodimers (r.m.s.d. < 0.41 A
˚
based on 7566 atoms). Although no activator is pre-
sent, the KlPDC tetramer contains an open and
a closed side and thus resembles more closely the
tetramer structure of pyruvamide-activated ScPDC
(Fig. 3) than that of the nonactivated ScPDC
(Fig. 4). In going from the nonactivated form of
ScPDC to the activated one, one dimer has to rotate
by about 30relative to the other. For comparison,
the corresponding angle found for (nonactivated)
KlPDC is 36. The main difference between KlPDC
and the activated form of ScPDC is the flexibility of
the loop regions 105–113 and 290–302. Whereas these
loops are completely ordered in pyruvamide-activated
ScPDC, residues 105–113 are completely disordered,
and 290–302 partially disordered, in KlPDC. As a
consequence, KlPDC resembles nonactivated ScPDC
more closely than activated ScPDC in terms of loop
flexibility.
Subunit structure
As in all other ThDP-dependent decarboxylases ana-
lyzed so far, the KlPDC subunit consists of three
domains (Fig. 2). According to Muller et al. [16], these
domains are termed the PYR domain (binding the am-
inopyrimidine ring of ThDP), the R domain (binding
regulatory effectors), and the PP domain (binding the
diphosphate residue of ThDP). All three domains exhi-
bit their typical a⁄b-topology. The central six-stranded
b-sheet of the PYR domain (residues 2–182) is sur-
rounded by seven a-helices. The R domain (residues
193–341) consists of five a-helices and a central six-
stranded b-sheet. A central six-stranded parallel
b-sheet and eight a-helices form the PP domain (resi-
dues 360–556). A superposition of ScPDC and KlPDC
monomers yields r.m.s.d. values < 0.85 A
˚(based on
3650 aligned atoms). The largest displacements are
observed for the C-terminal helix (5.5 A
˚) and for most
parts of the central R domain.
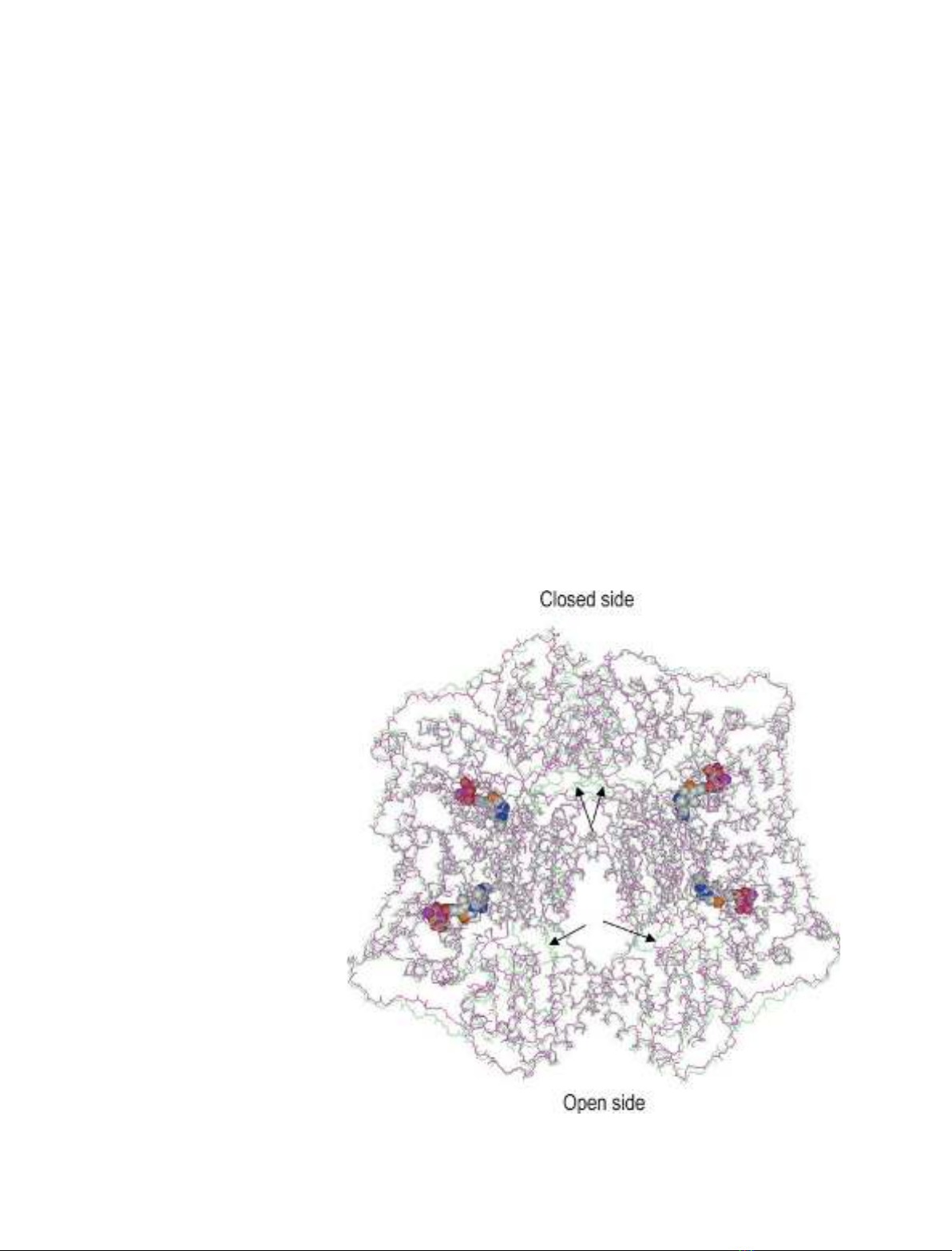
Fig. 3. Superposition of the main chain
atoms of tetramers of Kluyveromyces lactis
pyruvate decarboxylase (KlPDC) (pink) and
pyruvamide-activated Saccharomyces cere-
visiae PDC (ScPDC) (lime, PDB entry code
1QPB). The arrows indicate the loop regions
105–113 in each subunit, which are ordered
in pyruvamide-activated ScPDC and disor-
dered in KlPDC. The cofactors thiamine
diphosphate and Mg
2+
are shown in space-
filling mode. The closed and open sides of
the tetramers are indicated.
S. Kutter et al. Crystal structure of pyruvate decarboxylase
FEBS Journal 273 (2006) 4199–4209 ª2006 The Authors Journal compilation ª2006 FEBS 4203

























