
The solution structure of gomesin, an antimicrobial cysteine-rich
peptide from the spider
Nicolas Mandard
1
, Philippe Bulet
2
, Anita Caille
1
, Sirlei Daffre
3
and Franc¸oise Vovelle
1
1
Centre de Biophysique Mole
´culaire, CNRS, Orle
´ans, France;
2
Institut de Biologie Mole
´culaire et Cellulaire, CNRS, Strasbourg,
France;
3
Departamento de Parasitologia, ICB, Universidade de Sa
˜o Paulo, Brazil
Gomesin is the first peptide isolated from spider exhibiting
antimicrobial activities. This highly cationic peptide is
composed of 18 amino-acid residues including four cysteines
forming two disulfide linkages. The solution structure of
gomesin has been determined using proton two-dimensional
NMR (2D-NMR) and restrained molecular dynamics
calculations. The global fold of gomesin consists in a well-
resolved two-stranded antiparallel bsheet connected by a
noncanonical bturn. A comparison between the structures
of gomesin and protegrin-1 from porcine and androctonin
from scorpion outlines several common features in the
distribution of hydrophobic and hydrophilic residues. The
N- and C-termini, the bturn and one face of the bsheet are
hydrophilic, but the hydrophobicity of the other face
depends on the peptide. The similarities suggest that the
molecules interact with membranes in an analogous manner.
The importance of the intramolecular disulfide bridges in the
biological activity of gomesin is being investigated.
Keywords: spider; cysteine-rich; antimicrobial peptide;
bsheet; NMR.
In recent years, it has become widely recognized that animal
defense systems rely on inducible or constitutive expression
of antimicrobial peptides in response to bacterial and/or
fungal infections. Among these antimicrobial molecules,
open-ended cyclic cysteine-rich peptides are the most
widespread. They have been characterized in plants, inver-
tebrates and vertebrates. Structurally, they can be classified
into (a) peptides adopting a bsheet structure, namely the
mammalian defensins [1]; (b) peptides exhibiting the CSab
(cysteine stabilized ahelix bsheet) motif [2] such as defen-
sin A from Phormia terranovae [3], drosomycin from
Drosophila melanogaster [4], heliomicin from Heliothis
virescens [5], plant defensins [6]; and (c) peptides adopting
ab-hairpin-like fold, such as tachyplesins from horseshoe
crabs [7,8], porcine protegrins [9,10], thanatin from the bug
Podisus maculiventris [11], androctonin from the scorpion
Androctonus australis [12], lactoferricin B from bovine [13]
and a 20-residue antimicrobial peptide from the plant
Impatiens balsamina [14]. All the peptides adopting a
bhairpin structure possess a broad antimicrobial activity
spectrum. In contrast, peptides with a CSab motif have a
more restricted activity spectrum; insect defensins are
mainly active against Gram-positive bacteria whereas
drosomycin, heliomicin and plant defensins are active
exclusively against fungi.
While there are numerous reports on the structural
characterization and the three-dimensional structure of
polypeptide toxins from spider venoms (for review see [15]),
it is only very recently that a peptide with antimicrobial
activity has been characterized from spiders [16]. This
peptide, gomesin, is an 18-residue cysteine-rich antimicro-
bial peptide isolated from the blood cells (hemocytes) of the
mygalomorph spider Acanthoscurria gomesiana. Gomesin
has two disulfide bridges linking Cys2 to Cys15 and Cys6 to
Cys11. In addition, gomesin carries two post-translational
modifications: cyclization of the N-terminal glutamine into
pyroglutamic acid (pGlu or Z) and amidation of the
C-terminal arginine. The molecule is highly cationic
(pI ¼9.86 calculated by
EDITSEQ
from
DNA STAR
4.05
software) with the presence of five arginines, one lysine, a
C-terminal amidation and no acidic amino acid.
Gomesin exhibits broad activity at rather low concentra-
tions (often below 10 l
M
) against numerous microorgan-
isms including bacteria, filamentous fungi and yeast. In
addition, this peptide was found to affect the viability of the
parasite Leishmania amazonensis and to present some
hemolytic activity against human erythrocytes. Sequence
alignments suggest strong similarities with various anti-
microbial peptides adopting a bsheet structure, such as
tachyplesins, androctonin, and protegrins [16].
In this paper, we report on the elucidation of the solution
structure of gomesin using two-dimensional
1
H-NMR
spectroscopy and molecular modeling. Gomesin adopts a
well-defined b-hairpin-like structure as it could be expected
from the sequence similarities with androctonin and prote-
grins. The structure of these three peptides are compared in
order to determine the structural features required for their
biological properties.
MATERIALS AND METHODS
NMR experiments
Gomesin peptide was synthesized according to classical
Fmoc chemistry as described previously [16].
Correspondence to F. Vovelle, Centre de Biophysique Mole
´culaire,
CNRS UPR 4301, Rue Charles Sadron, 45071 Orle
´ans Cedex 2,
France. Fax: + 33 23863 1517, Tel.: + 33 23825 5574,
E-mail: vovelle@cnrs-orleans.fr
Abbreviations: pGlu (Z), pyroglutamic acid; PG-1, protegrin-1.
(Received 26 July 2001, revised 5 December 2001, accepted 2 January
2002)
Eur. J. Biochem. 269, 1190–1198 (2002) ÓFEBS 2002

The sample for NMR spectroscopy was prepared by
dissolving 4.5 mg of synthetic gomesin in 90%H
2
O/
10%D
2
O to obtain a final solution at 3.3 m
M
. The pH
was adjusted to 3.5 with microlitre increments of HCl 1 N.
For experiments in heavy water, 90% of the volume of the
previous sample was lyophilized and then dissolved in
99.99% D
2
O. The remaining volume (10%) was completed
with H
2
O to obtain a gomesin solution at 0.3 m
M
.
A conventional set of one-dimensional and two-dimen-
sional
1
H-NMR spectra in H
2
O, including DQF-COSY
[17], Clean-TOCSY [18] and NOESY [19], was acquired at
a temperature of 278 K on a VARIAN INOVA NMR
spectrometer equipped with a z-axis field-gradient unit and
operating at a proton frequency of 600 MHz. The clean-
TOCSY spectrum was collected with a spin lock time of
80 ms using the MLEV-17 mixing scheme [20] and
NOESY spectra were recorded with mixing times of
120 ms and 300 ms. Water suppression was achieved either
by presaturation for COSY and TOCSY experiments or
using the WATERGATE pulse sequence [21] for NOESY
experiments. A new series of TOCSY and NOESY spectra
in D
2
O was also recorded at 278 K. In an attempt to
overcome ambiguities in assignment due to spectral
overlap, a second set of clean-TOCSY and NOESY
spectra was performed at 285 K. Spectra were acquired
over a spectral width corresponding to 9 p.p.m. and
referenced to the residual H
2
O signal set as the carrier
frequency (4.964 p.p.m. at 278 K; 4.897 p.p.m. at 285 K).
All two-dimensional NMR data were processed on a
Silicon Graphics Indy O2 workstation using the
VNMR
software package (version 6.1; Varian, Inc., Palo Alto, CA,
USA). Assignments were carried out according to classical
procedures including spin-system identification and
sequential assignment [22] on maps recorded at 278 K.
Cross-peak intensities of the NOESY map at 278 K with
the shortest mixing time 120 ms and recorded over 4096
data points in the F2 dimension were integrated with
XEASY
[23].
The unusual N-terminal residue (pyroglutamic acid) was
especially built for this work and its coordinates and
appropriate parameters (bond length and atom charges)
were included in the libraries of
DYANA
[24,25] and
XPLOR
[26] for molecular modeling.
Structure calculations
NOESY cross-peak intensities were converted into upper
distance limit constraints using the
CALIBA
program [25].
The minimum distance constraint between two protons was
limited by their van der Waals radi (2.0 A
˚). Moreover, in
order to assess possible contributions from spin diffusion
effects, some NOEs only observable on the 300-ms mixing
time NOESY map were taken into account with a 6-A
˚
upper limit constraint. Each of the two disulfide bridges
was explicitly defined by three lower/upper distance limit
restraints between the sulphur and bcarbon atoms of
the two cysteines i, j involved in the linkage (1.9 A
˚<
d(Sc
i
,Sc
j
)<2.1 A
˚;2.5A
˚
<d(Cb
i
,Sc
j
)<3.5 A
˚;2.5A
˚<
d(Sc
i
,Cb
j
)<3.5 A
˚). All these constraints were brought
together in a distance restraint file used as input to initial
steps of molecular modeling. Several sets of 100 structures
were generated from random-built initial models using the
annealing procedure of the variable target function
program DYANA. During these rounds of calculations,
restraints corresponding to the stereospecific assignment of
three methyl protons proposed by
GLOMSA
were incorpor-
ated in the data set [25]. The hydrogen bonds found at each
round of calculations on a majority of structures and
corresponding to atoms involved in secondary structure
elements were also introduced as constraints. A final set of
50 structures was then generated in a final
DYANA
run from
an input file taking into account the total set of constraints.
Twenty out of these 50
DYANA
structures were selected on
the basis of low target function values (1A
˚
2
)and
subjected to energy minimization using Powell’s algorithm
and
CHARMM
force field parameters [27] implemented in
X
-
PLOR
3.1 software. The energy calculations were
performed with a distance dependent dielectric function
e¼r,a12-A
˚cut-off distance for all nonbonded interac-
tions and a force constant of 50 kcalÆmol
)1
ÆA
˚
)2
for NOE
restraint energy terms. All calculations were carried out on
a Silicon Graphics 02 R10000 workstation and the struc-
tures were visualized with the
SYBYL
software (TRIPOS
Inc., St Louis, MO, USA). Hydrophobic potentials were
calculated with the
MOLCAD
option [28] implemented in
SYBYL
.
PROCHECK
[29] and
PROMOTIF
[30] programs were
used for structural analysis.
RESULTS AND DISCUSSION
Sequence-specific assignment and secondary structure
Comparison of the one-dimensional spectra of the samples
of gomesin at 0.3 m
M
and 3.3 m
M
in aqueous solution
clearly shows the absence of any concentration-dependent
changes in the chemical shifts or peak line widths, suggesting
the monomeric state of the peptide in our experimental
conditions. The two-dimensional
1
H-NMR spectra of
gomesin were assigned via standard sequential assignment
methods developed by Wu
¨thrich [22]. The entire spin
systems of individual amino-acid residues were identified
through DQF-COSY and TOCSY experiments on the
maps at 278 K. TOCSY and NOESY maps recorded at
285 K were used to clear up ambiguities in the assignment
of the NH-Hacross-peaks of Arg4 due to the close vicinity
of its Hachemical shift and of the water resonance.
Moreover, dipolar connectivities on the D
2
O NOESY
spectra enable the best-defined Ha-Hapeaks to be obtained
near the residual water diagonal, especially between Cys2
and Cys15, Cys6 and Cys11, Arg4 and Thr13. The splitting
of the resonance of backbone NH and Haprotons allows
complete proton assignments for the fingerprint region
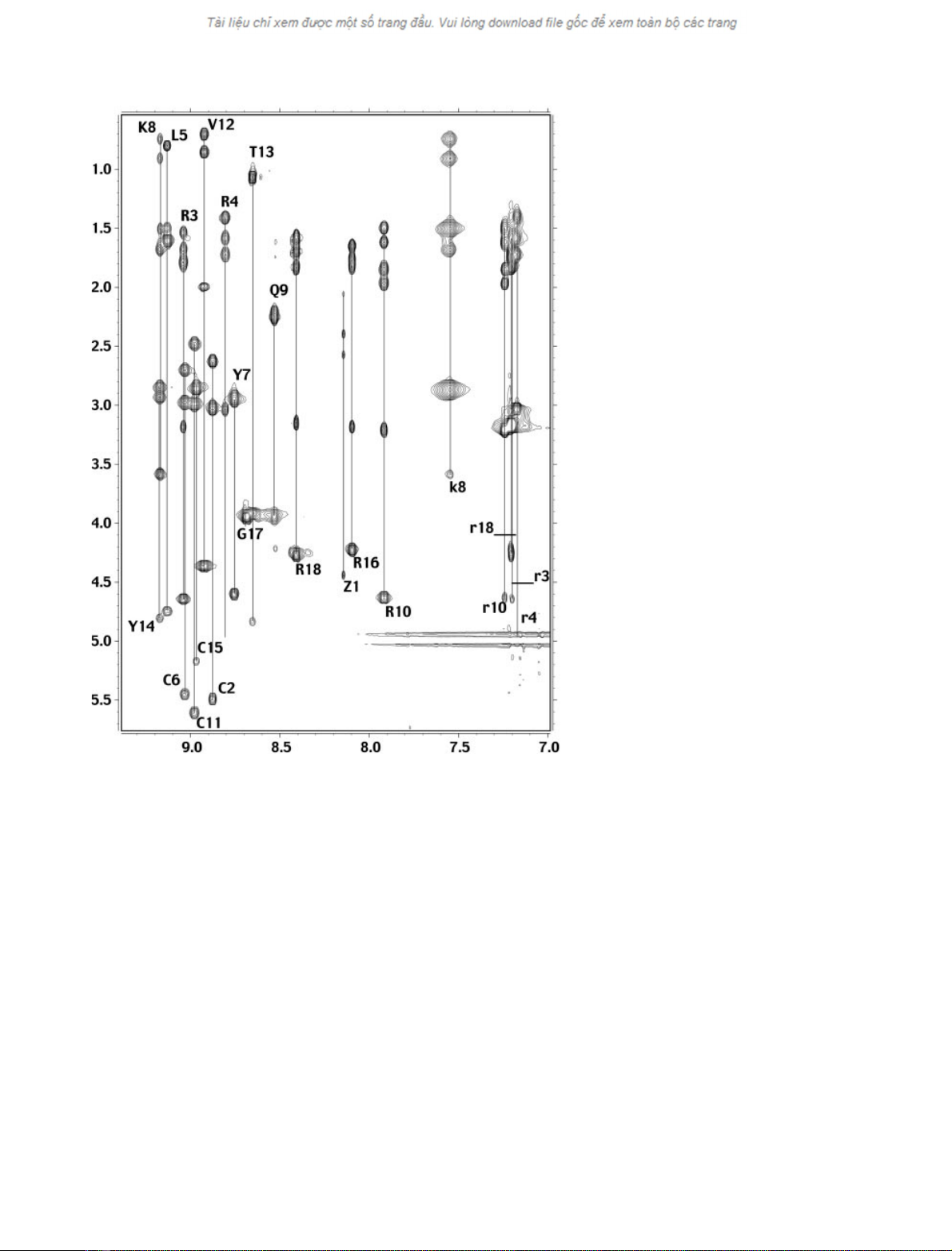
(Fig. 1).
1
H chemical shifts of gomesin are reported in
Table 1 and the complete pathway Ha(i))NH(i+1)is
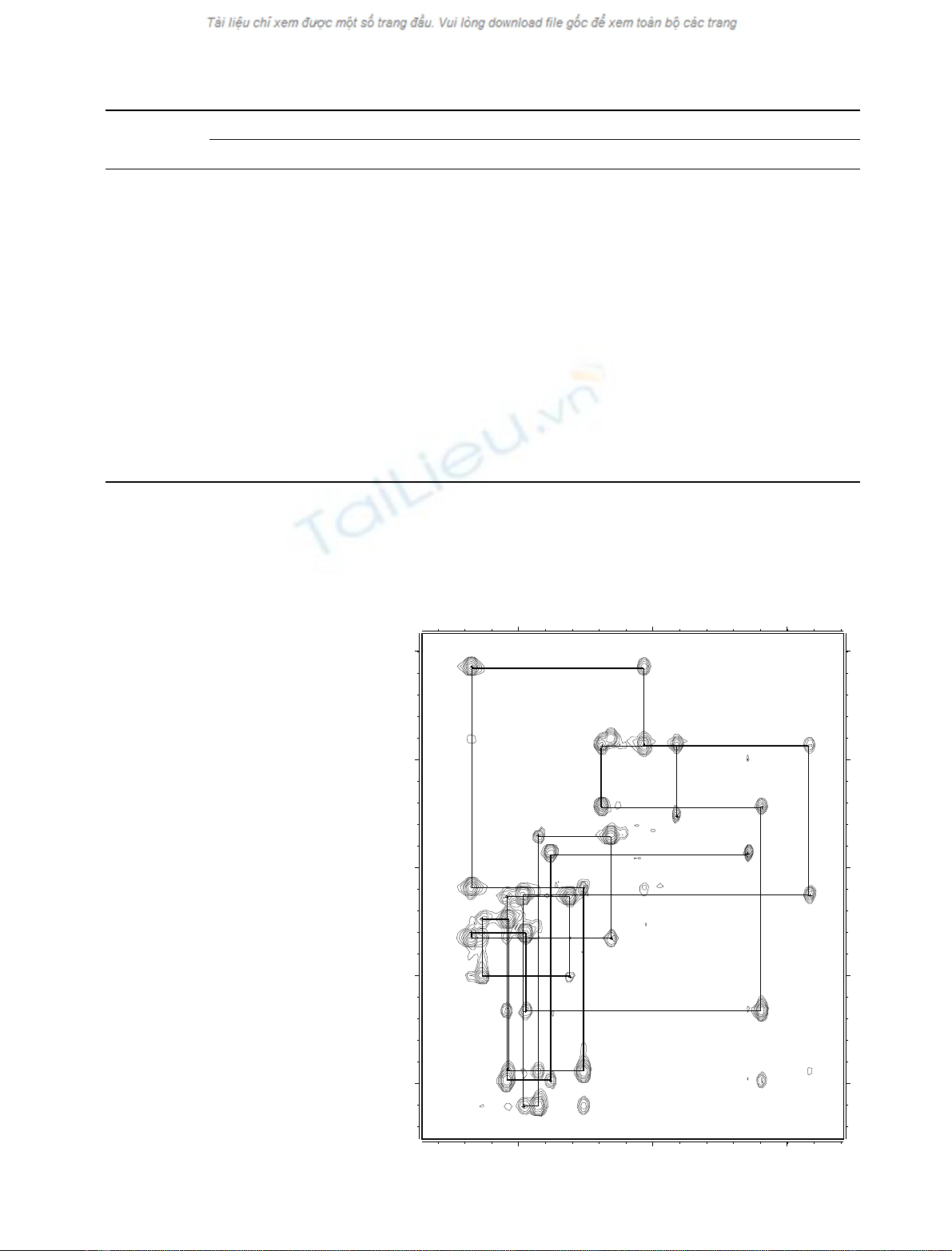
shown in Fig. 2. The NOE connectivity diagram exhibits
d
NN
(i,i+2)andd
aN
(i,i+ 2) NOEs between the central
residues (Tyr7–Arg10), suggesting the presence of a turn in
this region (Fig. 3A). Strong d
aN
(i,i+1)NOEsinseg-
ments (Cys2–Cys6 and Cys11–Cys15) are indicative of two
extended strands of bsheet. This hypothesis is confirmed by
the presence of long-distance Ha(i)-Ha(j) connectivities
detected on D
2
O maps even if deuterium exchange studies
revealed that all amide protons were quickly exchanging
with the solvent. Figure 3B shows the number of NOEs
between two residues i and j with respect to the difference
|i)j|. The enhancement of the number of NOEs observed
ÓFEBS 2002 Solution structure of gomesin (Eur. J. Biochem. 269) 1191
for 5<|i)j|<13 is mainly due to connectivities between
the protons of residues Cys6 and Cys11 (|i)j|¼5), Leu5
and Val12 (|i)j|¼7), Arg4 and Thr13 (|i)j|¼9), Arg3
and Tyr14 (|i)j|¼11), Cys2 and Cys15 (|i)j|¼13).
Finally, no NOE cross-peak, indicative of an oligomeric
association in solution, could be detected, which is consis-
tent with the high abundance of positively charged residues
(five arginines and one lysine) in the primary structure of the
peptide.
Structure evaluation
The three-dimensional structure of gomesin was determined
using the standard simulated annealing protocol of
DYANA
AND
energy minimization with
X
-
PLOR
, as described in
Materials and methods. The final restraint file comprised a
set of 289 distance restraints including 82 intraresidual, 102
sequential, 32 medium-range (2 < |i)j| < 5) and 73 long
range (|i)j|‡5) restraints (with an average of 16 restraints
per residue). Long-range limits concern mainly residues
located in the segments corresponding to the two strands of
the bsheet (pGlu1–Tyr7; Arg10–Arg16) (data not shown).
As shown in Table 2, the 20 selected structures are in very
good agreement with all experimental data and the standard
covalent geometry. There are no violations larger than
0.3 A
˚and the root-mean-square deviations (rmsd) with
respect to the standard geometry are low. Both negative van
der Waals and electrostatic energy terms are indicative of
favorable non-bonded interactions. Moreover, the Rama-
chandran plot exhibits nearly 91% of the (/,w) angles of all
structures in the most favored regions and additional
allowed regions according to the
PROCHECK
software
nomenclature. The structure files have been deposited at
the Protein Data Bank (http://www.rcsb.org/pdb) with the
accession number 1KFP.
Structure description
The overall fold of gomesin is formed by a hairpin-like
structure with a two-residue extension at the C-terminal
end. This hairpin-like structure consists of two antiparallel
bstrands (pGlu1–Tyr7 facing Arg10–Arg16) forming a
twisted sheet and connected by a four-residue turn (Tyr7–
Arg10). As shown in the structural statistics (Table 2) and
Fig. 1. Fingerprint region of a TOCSY spec-
trum of gomesin in 90%H
2
O/10%D
2
Oat
5°C, pH 3.5. The spin systems of the amide
protons are designated by the amino acid one-
letter code, upper case letters. The spin system
of side chain nitrogen-bond protons is indi-
cated with the amino acid one-letter, lower
case letters.
1192 N. Mandard et al. (Eur. J. Biochem. 269)ÓFEBS 2002
by superimposition of the 20 structures (Fig. 4), the
structures are extremely well defined. The pairwise rmsd
on the N, Ca,C¢backbone atoms of residues 1–16 is only
0.34 A
˚and drops to 0.17 A
˚when calculated in the bsheet
region. Several main structural elements contribute to a
strong stabilization of the sheet. Six regular backbone-
backbone hydrogen bonds characteristic of the bsheet
structure, NH(Arg3)–O(Tyr14), O(Arg3)–NH(Tyr14),
Table 1.
1
H chemical shifts (p.p.m.) for gomesin in aqueous solution at 278K, pH 3.5.
Residue
Chemical shifts
NH HaHbOthers
pGlu1 8.15 4.44 2.40, 2.05 Hc2.57, 2.57
Cys2 8.88 5.48 3.02, 2.63
Arg3 9.04 4.64 1.79, 1.69 Hc1.54, 1.54; Hd3.18, 3.18; NHe7.20
Arg4 8.80 5.00 1.73, 1.58 Hc1.42, 1.42; Hd3.03, 3.03; NHe7.18
Leu5 9.13 4.74 1.60, 1.60 Hc1.51; Hd0.81, 0.81
Cys6 9.03 5.44 2.98, 2.70
Tyr7 8.76 4.59 2.94, 2.94 Hd7.15; He6.78
Lys8 9.17 3.58 1.69, 1.69 Hc0.91, 0.75; Hd1.51, 1.51; He2.88, 2.88; NHe7.56
Gln9 8.53 3.94 2.21, 2.21 Hc2.25, 2.25
Arg10 7.92 4.63 1.97, 1.85 Hc1.61, 1.50; Hd3.21, 3.21; NHe7.24
Cys11 8.98 5.60 2.99, 2.48
Val12 8.92 4.35 2.00 Hc0.86, 0.71
Thr13 8.65 4.83 3.91 Hc1.07
Tyr14 9.17 4.80 2.94, 2.85 Hd7.05; He6.73
Cys15 8.97 5.16 2.86, 2.86
Arg16 8.10 4.22 1.83, 1.75 Hc1.65, 1.65; Hd3.18, 3.18; NHe7.21
Gly17 8.69 3.94, 3.94
Arg18 8.41 4.27 1.84, 1.70 Hc1.59, 1.59; Hd3.15, 3.15; NHe7.21
8.08.59.0
3.5
4.0
4.5
5.0
5.5
11
6
15
2
14 53
12
7
13
8
17 9
18 1
16
10
4
Fig. 2. Amide-aregion of a 120-ms mixing
time NOESY spectrum of gomesin. For the
sake of clarity, only the intraresidue a-amide
cross-peaks are labeled.
ÓFEBS 2002 Solution structure of gomesin (Eur. J. Biochem. 269) 1193

NH(Leu5)–O(Val12), O(Leu5)–NH(Val12) are found
between the disulfide bridges as well as O(pGlu1)–
NH(Arg16) and NH(Tyr7)–O(Arg10) located at each
extremity of the bsheet. Two interstrand disulfide bridges
adopt a well-defined right-handed conformation with v
SS
,
v
1
,v
2
torsion angles close to the expected values for
favorable energy conformers (Table 2). Moreover, whatever
the model considered, the average distance between the Ca
atoms of the cysteine residues is small (3.75 ± 0.10 A
˚). This
often occurs when disulfide bridges link antiparallel
b-strands [31]. The backbone of the loop (Tyr7-Lys8-
Gln9-Arg10) also exhibits a well-defined conformation.
When the structures are best fitted on the four backbone
residues of the turn, the local pairwise rmsd of this turn is
0.22 A
˚. The (i,i+ 3) hydrogen bond between the CO group
of Tyr7 and the NH group of Arg10 closing classical bturns
is found only on 10 out of the 20 structures. Whatever the
nomenclature used ([32] or [33]), this turn appears to be
particularly difficult to classify as Lys8 exhibits positive /
and wangles as observed in a left-handed helix and the /,w
average values ()150°,–60°)ofGln9areveryunusual.
Owing to a lack of NOE data, the conformation of the two
C-terminal residues Gly17 and Arg18, which are not
included in the bsheet, is poorly defined.
Most side chains of strand residues adopt a well-defined
conformation due to the presence of numerous interstrand
NOEs. In particular, significantly low circular variances [33]
for v
1
and v
2
angles are observed for the four cysteines, for
Tyr7, Val12, Thr13 and Tyr14 residues (CV < 0.1). Low v
1
and v
2
circular variances are also observed for long chain or
bulky residues such as Arg4, Leu5, and Arg10 but, in these
cases, the extremity of their side chain is rather floppy. In
contrast, the side chains of Arg16 and Arg18 at the
Table 2. Structural statistics of the 20 models of gomesin. Ramachandran plots were calculated with
PROCHECK
and the energy terms were calculated
using the
CHARMM
force field.
Restraint violations, mean number per structure (min, max)
Distance restraints > 0.3 A
˚0.7 (0, 2)
Distance restraints > 0.2 A
˚1.6 (1, 4)
Deviation from standard geometry, mean number per structure (min, max)
Bond lengths > 0.05 A
˚0.3 (0, 1)
Bond angles > 10°0.2 (0, 2)
Ramachandran Maps (%)
Most favourable regions 77.0
Additional regions 13.7
Cysteine side chain torsion angles (average values in degrees)
i)jvi
1vi
2v
SS
vj
2vj
1
Cys2-Cys15 )60.3 ± 3.1 )84.4 ± 4.5 103.6 ± 3.0 )84.7 ± 3.5 )70.7 ± 3.5
Cys6-Cys11 )65.7 ± 2.5 )96.0 ± 3.2 96.0 ± 1.4 )70.6 ± 2.7 )67.7 ± 3.4
Final energies (kcalÆmol
)1
)
E
total
)163 ± 11
E
electrostatic
)251 ± 11
E
vdw
)50.0 ± 2.5
E
NOE
13.2 ± 2.0
Average rmsd (N-Ca-C¢) Pairwise (A
˚) Mean structure (A
˚)
Whole 0.79 ± 0.32 0.51 ± 0.19
Hairpin 0.34 ± 0.08 0.24 ± 0.07
bsheet 0.17 ± 0.07 0.14 ± 0.05
Turn 0.22 ± 0.10 0.15 ± 0.07
A
dNN(i,i+1)
dαN(i,i+1)
dβN(i,i+1)
dNN(i,i+2)
dαN(i,i+2)
dαN(i,i+4)
5
Z CRRL CYKQ10
RC
VT Y15
CRGR
0 4 8 12 16
0
20
40
60
80
100
120
Range |i-j|
Number of NOEs
B
Fig. 3. NOE connectivities and number. (A) Summary of the sequential
NH(i))NH(i+1), Ha(i))NH(i+1), Hb(i))NH(i+1), and
medium range NH(i))NH(i+2), Ha(i))NH(i+2), Ha(i) )
NH(i+ 4) connectivities identified for gomesin. The height of the bars
reflects the strength of the NOE correlation as strong, medium and
weak. (B) Number of NOEs vs. difference |i)j|.
1194 N. Mandard et al. (Eur. J. Biochem. 269)ÓFEBS 2002

























