
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/316965450
Current methods of extracellular DNA methylation analysis
ArticleinMolecular Biology · March 2017
DOI: 10.1134/S0026893317010071
CITATIONS
8
READS
1,993
2 authors:
Olga E Bryzgunova
Institute of Chemical Biology and Fundamental Medicine, Russian Academy of Sci…
66 PUBLICATIONS1,762 CITATIONS
SEE PROFILE
Pavel Petrovich Laktionov
Russian Academy of Sciences, SD, Novosibirsk
278 PUBLICATIONS5,591 CITATIONS
SEE PROFILE
All content following this page was uploaded by Olga E Bryzgunova on 21 March 2018.
The user has requested enhancement of the downloaded file.

167
ISSN 0026-8933, Molecular Biology, 2017, Vol. 51, No. 2, pp. 167–183. © Pleiades Publishing, Inc., 2017.
Original Russian Text © O.E. Bryzgunova, P.P. Laktionov, 2017, published in Molekulyarnaya Biologiya, 2017, Vol. 51, No. 2, pp. 195–214.
Current Methods of Extracellular DNA Methylation Analysis
O. E. Bryzgunovaa, b, * and P. P. Laktionova, b
aInstitute of Chemical Biology and Fundamental Medicine Siberian Division
Russian Academy of Sciences, Novosibirsk, 630090 Russia
bE.N. Meshalkin Siberian Federal Biomedical Research Center, Ministry of Health Care of Russian Federation,
630055, Novosibirsk, Russia
*e-mail: olga.bryzgunova@niboch.nsc.ru
Received February 27, 2016; in final form, March 21, 2016
Abstract⎯The discovery of the enormous role methylated cytosine plays in regulating gene expression has led
to the development of a variety of techniques for detecting cytosine modification. A majority of these tech-
niques are geared towards analyzing genomic DNA, which is typically available in large quantities. The con-
centration of cell-free DNAs (cfDNA) extracted from biological fluids including plasma, saliva, tears, or
urine is relatively low and their degree of the fragmentation is high. Moreover, for noninvasive diagnostics of
cancer, methylation patterns must be studied in minor cancer-specific fractions of DNA molecules substan-
tially diluted by excess unmethylated molecules. The above limitations complicate the application of tradi-
tional techniques for cfDNA methylation analysis. In this manuscript, we review the state-of-art analysis of
cfDNA methylation, hydroxymethylation, and noncanonical methylation (outside of CpG islands). The
review covers methodological approaches to studying individual CpGs and genomic loci, as well as tech-
niques for the large-scale analysis of methylation.
Keywords: methylation, hydroxymethylation, non-CpG methylation, cell-free DNA, restriction endonucle-
ase, emulsion PCR, microarray
DOI: 10.1134/S0026893317010071
INTRODUCTION
Methylation of cytosines in the 5'-position of the
pyrimidine ring is a physiological process that is active
in all normal cells of the body. The proportion of
5-methylcytosine in the human genome is about 1% of
all bases and varies slightly in different types of tissues
with 75% of CpG dinucleotides being methylated [1].
It is known that at least 10% of expressed genes are
regulated by methylation and up to 60% of genes have
local CpG islands [2]. Changing the methylation sta-
tus of the promoter regions of genes involved in onco-
genesis including tumor suppressor genes may lead to
the inactivation (methylation) or activation (demeth-
ylation) of their expression and may promote the
emergence and proliferation of tumor cells. Increased
methylation status that is not detected in the norm is
revealed by the PCR analysis of genes in blood plasma
or serum in pancreatic cancer (Р16, 24.6% of cases)
[3], prostate cancer (RASSF1, RAR
β
2, GSTP1; 24, 12,
and 13%, respectively) [4], breast cancer (АРС and
GSTP1, 17 and 26%, respectively) [5], hepatocellular car-
cinoma (P16, 19–81%) [6, 7], ovarian cancer (in the
simultaneous analysis of six genes, i.e., BRCA1, RASSF1A,
APC, P14arf, P16ink4a, and DAPK, with 100% specific-
ity and 82% sensitivity [8]), and many others. Some
epigenetic changes (gene hypermethylation) is known
to occur at early stages (IA or B) of cancer [9, 10].
That is why the study of the methylation status of
the genes in cell-free DNA (cfDNA) is an important
task in modern biology. Every year, a growing number
of studies are devoted to analyzing the aberrant meth-
ylation of cytosines in cfDNA of various biological
fluids because this DNA is a convenient source of
diagnostic material for developing systems for early
noninvasive diagnostics and monitoring anticancer
therapy. Not all methodological approaches to study-
ing the methylation of genomic DNA can be applied to
analyzing aberrant methylation in cfDNA. Their use is
limited by the low content of cfDNA (up to 53 ng/mL
and 10–2160 ng/mL in the blood plasma of healthy
Abbreviations: cfDNA, cell-free DNA; MPS, massively parallel
sequencing; BLM-PCR blunt-ended ligation-mediated PCR;
HPLC, high-performance liquid chromatography; MALDI-
TOF MS, matrix-assisted laser desorption/ionization, time-of-
flight mass spectrometry; MeDIP, methylated DNA immuno-
precipitation; MethDet56, microarray-mediated methylation
analysis of 56 fragments in each sample; Methyl-BEAMing,
beads, emulsion, amplification, and magnetics; MIRA, methyl-
ated CpG island recovery assay using MBD2 proteins; MSP,
methylation specific PCR; Ms-SNuPE, methylation-sensitive
single nucleotide primer extension; qMAMBA, quantitative
methylation analysis of minute DNA amounts after whole bisul-
fitome amplification); SMRT, single molecule real-time
sequencing); tSMS, true single molecule sequencing
REVIEWS
UDC 57.088.1

168
MOLECULAR BIOLOGY Vol. 51 No. 2 2017
BRYZGUNOVA, LAKTIONOV
donors [11–15] and in cancer patients [13, 16–18],
respectively, and from 2 ng/mL to 96 ng/mL in the
urine of healthy donors and cancer patients [19–21].
Moreover, as a rule, cfDNA is low molecular DNA;
the size of cfDNA fragments in the blood plasma and
serum of healthy and sick people is most often 100–
200 bp, although there is high molecular cfDNA with
sizes of up to 10000 bp; the sizes of cfDNA fragments
in the urine of men and women are 150–250 bp [16–
20, 22–28]. It should be noted that the content of
tumor DNA in the blood of cancer patients is 1–2% of
the total pool of cfDNA [22]; there is little information
that the content of tumor DNA can achieve 93% [16].
The small amount of cfDNA complicates its analysis
and requires special methodology.
Thus, the methods of studying the methylation of
cytosines in cfDNA must meet the following require-
ments: they should make it possible to work with a
minimum amount of mostly short DNA fragments
and to analyze methylation of minor pool of DNA
molecules (DNA from cancer cells) in the presence of
the excess of ballast DNA (from normal tissues).
Obviously, this task cannot be solved without amplify-
ing initial DNA. For this purpose, both the PCR of
specific predefined loci and the whole-genome ampli-
fication of cfDNA samples may be used. The applica-
tion of the amplification methods for analyzing
cfDNA is determined by the methodology of the sub-
sequent analysis of DNA. These methods will be
described in the corresponding chapters.
All approaches to studying the methylation of both
genomic and cell-free DNA may be divided into three
groups as follows:
(1) chemical methods, which are based on the
selective modification of methylated and nonmethyl-
ated cytosines;
(2) enzymatic methods, which are based on the use
of restriction endonucleases sensitive to m5С in the
recognition site;
(3) mixed methods, which are the combination of
the above approaches.
All methods are used to solve two classes of prob-
lems, i.e., the study of the methylation of selected loci
or methylation on the level of the whole genome. Two
chapters of this review describe the methodological
approaches that are used for solving the tasks of the
first type; the separate chapter describes the methods
of the large-scale study of methylation of extracellular
DNA.
IDENTIFICATION OF METHYLATED
CYTOSINES WITHOUT USING
METHYLATION-SENSITIVE RESTRICTION
ENDONUCLEASES
The method for analyzing the cytosine methylation
status that was developed over 20 years ago is based on
the modification of cytosine with bisulfite [29]. The
studied sample (cfDNA in our case) is treated with
sodium bisulfite, which leads to the complete sulfona-
tion of cytosines without the m5C modification. Sub-
sequent desulfonation under alkaline conditions leads
to the deamination of cytosines and their conversion to
uracils. It is clear that after the conversion and amplifi-
cation, m5C and nonmethylated cytosine are deter-
mined as cytosine [30, 31] and thymine, respectively.
At the next stage, one or both DNA strands are
amplified using primers (containing or not containing
CpG dinucleotides) complementary to converted
DNA (after the deamination of nonmethylated cyto-
sines, DNA loses its ability to complementary interac-
tions). After bisulfite conversion, it is possible to
increase the amount of cfDNA by the whole-genome
amplification as was performed, e.g., in the qMAMBA
method (quantitative methylation analysis of minute
DNA amounts after whole bisulfitome amplification
[32]). The authors believe that this approach makes it
possible to study methylation starting from 100 pg of
DNA. The resulting PCR products were sequenced [31],
pyrosequenced [33], cloned in vector, and the inser-
tions are sequenced [34] or the sequencing was per-
formed by methods of mass parallel sequencing
(MPS) [35]. The use of methylation independent
primers, which are complementary to the regions con-
taining no CpG dinucleotides, makes it possible to
determine the composition and concentration of
DNA. In the case of low amounts of DNA, whole
genome amplification methods are when direct
amplification is impossible or when DNA methylation
is studied by methods such as microarray analysis or
MPS. The advantages of the bisulfite conversion
method include its high sensitivity (from 2 pg of DNA
[36]), the accuracy due to the simultaneous analysis of
initial complementary strands, and the possibility to
study the methylation status of individual DNA mole-
cules. The drawbacks of this method are partial DNA
degradation during modification/deamination [37],
incomplete DNA denaturation in the regions of
extended repeats or complicated secondary structure,
and the incomplete modification of cytosines or
incomplete deamination. These drawbacks may be
minimized by optimizing conditions for the bisulfite
conversion. It should be noted that modern commer-
cial kits for the bisulfite conversion of cfDNA, e.g.,
Zymo Research (United States) and Qiagen (Ger-
many) allow one to efficiently perform this reaction in
a high yield of target DNA [33].
One of the examples is bisulfite sequencing using
primers containing no CpG dinucleotides. This
method allows one to analyze the aberrant methyla-
tion of individual cytosines in cfDNA from biological
fluids [31]. Isolated cfDNA is initially treated with
sodium bisulfite; PCR is then carried out with primers
containing no CpG dinucleotides; the resulting PCR
products are purified by electrophoresis in polyacryl-
amide gel to sequence the desired PCR product. The

MOLECULAR BIOLOGY Vol. 51 No. 2 2017
CURRENT METHODS OF EXTRACELLULAR DNA 169
purified PCR products are sequenced by the Sanger
method using forward and reverse primers. The
advantage of analyzing DNA methylation by Sanger
sequencing is the possibility of studying extended
sequences, while a disadvantage of this method is the
inability to quantify the results.
After bisulfite conversion, cfDNA is analyzed by
the pyrosequencing method, which is based on detect-
ing pyrophosphate released during the synthesis of the
second DNA strand on the studied single-stranded
DNA template. To enhance the sensitivity of the
method, PCR is often used in two stages (nested
PCR) using primers containing no CpG dinucleo-
tides. At the second stage, one biotinylated primer is
used for the isolation of the sequenced strand. The sin-
gle-stranded biotinylated DNA is isolated using sep-
harose beads covered with streptavidin, followed by
pyrosequencing on a PSQ 96MA pyrosequencer using
sequencing primer and the PyroGold SQA reagent kit
(Pyrosequencing AB, Sweden) [33]. On a model sys-
tem, the authors showed that amplification does not
influence the ratio of methylated and unmethylated
DNA. It should be noted that the analysis of DNA by
pyrosequencing can be carried out on 96-, 48-, and
24-well platforms (www.qiagen.com).
After bisulfite conversion, aberrant methylation of
cytosines in cfDNA in the presence of 97–99% of
unmethylated DNA may be detected by one of the
PCR variants. Methylation-specific PCR (MSP) uses
two pairs of primers, one of which is specific to bisul-
fite-converted methylated DNA target sequences, and
the other is specific to unmethylated counterpart
sequences of bisulfite-treated DNA. Thus, the former
primers are only attached to regions containing methyl-
ated cytosines, while the latter primers are only attached
to unmethylated cytosine-containing regions. The
other variant of PCR is Combined Bisulfite Restriction
Analysis (COBRA). Endonucleases, such as BstUI, are
used for restriction. The results of MSP and COBRA
are analyzed by electrophoresis in a polyacrylamide
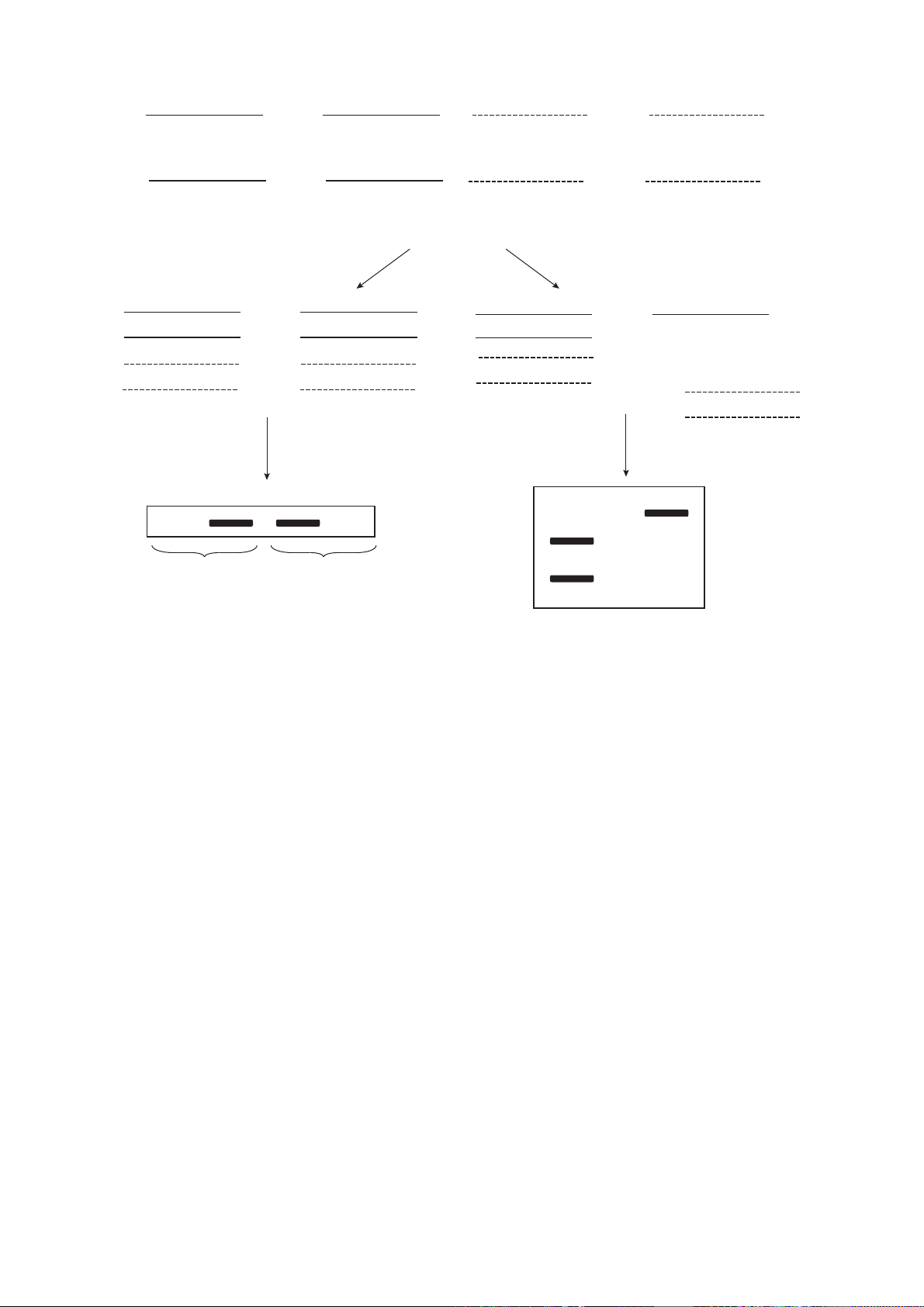
gel (Fig. 1) [38–41]. Another variant of the analysis of
methylated cfDNA is quantitative TaqMan or
SybrGreen I PCR using primers/probes that are com-
plementary to one of the converted and initially meth-
ylated strand, i.e., a strand containing CpG dinucleo-
tides, and unmethylated DNA strand [42].
The Methyl-BEAMing (beads, emulsion, amplifi-
cation, and magnetics) PCR method allows one to
detect methylated cfDNA in the presence of 99.9% of
the unmethylated form (Fig. 2) [43]. In this case,
cfDNA is initially hydrolyzed by nucleases, diluted to
the concentration of several nanograms in a milliliter,
and subjected to bisulfite conversion. The resulting
individual cfDNA molecules are amplified in an
emulsion of so-called water nanoparticles in a contin-
uous oil phase. Each water nanoparticle contains
DNA polymerase, the necessary cofactors, dNTP,
one DNA template molecule, and a DNA-binding
magnetic bead. Thus, each bead binds thousands of
identical copies of one DNA template molecule within
its own water nanoparticle. This process is similar to
the cloning of individual DNA fragments into a plas-
mid vector with the formation of the bacterial colony.
After PCR completion, the oil emulsion is destroyed
and the beads are collected. The individual status of
each DNA molecule is estimated by hybridization of
resultant PCR products with fluorescent probes that
specifically bind to initially methylated or unmethyl-
ated DNA sequences. The results are analyzed by flow
cytometry.
The Methyl-BEAMing method is similar to ana-
lyzing methylated cfDNA by digital PCR when the
reaction mixture is divided into a plurality of microre-
actors and one or several DNA molecules are ampli-
fied in each of them. This amplification can be carried
out using the commercial QX100TM Droplet DigitalTM
PCR system (Bio-Rad), which makes it possible to
work with small amounts of cfDNA (0.5 mL of blood
plasma) without molecular standards and ethalon
[44]. The analysis of the methylation status of cyto-
sines in cfDNA by digital PCR may be performed
using the commercial BioMark System (Fluidigm) on
platform 12.765 Digital Arrays (Fluidigm) containing
12 panels of 765 reaction chambers each. This system is
used to study fetal cfDNA in the maternal blood [45].
Another method suitable for identifying aberrantly
methylated cytosines in cfDNA after chemical conver-
sion is MethyLight PCR (Fig. 3а) [46]. Probes con-
taining different fluorescent labels specifically bind to
methylated or unmethylated DNA regions. This
method makes it possible to identify methylated DNA
in the presence of a 10000-fold excess of unmethylated
DNA [47].
The variant of HeavyMethyl PCR is also applicable
to analyzing the cytosine methylation status in cfDNA
(Fig. 3b). This method uses methylation-specific oli-
gonucleotide blockers and probes for the methylation-
specific amplification and detection of DNA [48]. The
amount of 30 pg of methylated DNA is sufficient for
analysis in the presence of 50 ng of unmethylated
DNA. The principle of the HeavyMethyl method is as
follows. Blocker oligonucleotides cannot bind to
methylated DNA, and this region is free to bind to
primers (gray arrows), thus making amplification of
DNA possible in this case. The fluorescent methyla-
tion-specific probe provides visualization of the sig-
nal. In the case of unmethylated DNA (Fig. 3b),
blocker oligonucleotides bind to DNA and prevent the
access of primers, thus inhibiting amplification.
One of the disadvantages of the MethyLight,
HeavyMethyl, and other PCR variants that use bisul-
fite-converted DNA is the inability to assess the meth-
ylation of individual cytosines in the studied DNA
region. However, this problem is solved by combining
the methods of HeavyMethyl PCR and methylation-
sensitive single nucleotide primer extension (Ms-
170
MOLECULAR BIOLOGY Vol. 51 No. 2 2017
BRYZGUNOVA, LAKTIONOV
SNuPE) [49]. This analysis requires 14 pg of DNA,
and aberrantly methylated sites can be identified in the
sample with a 2000-fold excess of normal DNA. The
principle of the Ms-SNuPE method is the amplifica-
tion of the studied region after bisulfite conversion of
DNA using primers specific to converted DNA [50].
The resulting PCR products are incubated with the
Ms-SNuPE primer(s), which are designed to be
hybridized just before cytosine that should be investi-
gated for methylation. Incubation is carried out in a
buffer containing [32Р]dСTP, [32Р]dТTP or fluores-
cently labeled dCTP/dTTP, and Taq polymerase. The
labeled products are visualized after electrophoresis in
denaturing polyacrylamide gel. The methylation status
is determined by the base that is incorporated in the
polymer chain. It was suggested to use ddCTP/ddTTP
or α-S-ddCTP/α-S-ddTTP instead of dСTP/dТTP and
to assess the incorporation of a nucleotide by high perfor-
mance liquid chromatography (HPLC) [51] or by matrix-
assisted laser desorption/ionization and time-of-flight
mass spectrometry (MALDI-TOF-MS) [52, 53].
The methylation status of individual cytosines in
bisulfite-converted cfDNA is analyzed by the combi-
nation of the HeavyMethyl and Ms-SNuPE methods,
with the former being followed by the latter. The
results are analyzed by HPLC [49].
It is clear from the above that the modern methods
allow for the successful identification of the cfDNA
methylation status in biological fluids, including in the
presence of a significant (up to 2000-fold) excess of
unmethylated DNA.
IDENTIFICATION OF METHYLATED
CYTOSINES IN DNA BY METHYLATION-
SENSITIVE RESTRICTION ENDONUCLEASES
Another approach that makes it possible to detect the
cytosine methylation status of specific DNA sequences is
fundamentally different from the above methods. This is
based on the use of restriction endonucleases that are
sensitive or insensitive to methylated cytosines. Both the
individual enzymes (e.g., Bsh1236I (BstUI) [54] and
Tsp509I [55]) and the pairs of endonucleases (e.g.,
SmaI/XmaI and HpaII/MspI [56, 57]) can be used for
this purpose. The most commonly used pairs are
MspI/HpaII, which recognizes the CCGG sequence
(HpaII is sensitive to methylation of the second cyto-
sine in the site) [58] and HpaII/HinP1I that recog-
Fig. 1. Concept of methods of MSP (methylation specific PCR) and COBRA (combined bisulfite restriction analysis) for ana-
lyzing the cytosine methylation status.
CC
CC
GG CCGG
CCGG
CCGG
GG
Bisulfite
treatment
TT
GGTT
Unmethylated DNA (Unmet) Methylated DNA (Met)
Unmethylated
DNA
Methylated
DNA
PCR with primers
specific for methylated
or unmethylated DNA
AA
MSP COBRA
Electrophoresis
Met
met
met
MetUnmet Unmet
PCR with primers
containing no CpG
dinucleotides + restriction
endonuclease BstUI
+
Met Unmet
CCGG
met
met
C
CG
G
CG C
GGTT
A
CG





![Nấm da mặt: Đặc điểm lâm sàng, cận lâm sàng [chuẩn nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20251030/vitobirama/135x160/43151768450819.jpg)

![Đánh giá cập nhật về sản xuất và ứng dụng Probiotic [năm hiện tại]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20250919/trungnguyen1718246@gmail.com/135x160/73411758335307.jpg)
















![Giáo trình Thực tập Vi sinh y học [Chuẩn Nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260325/viellison-95/135x160/2791774417404.jpg)
