
A mitochondrial cytochrome bmutation causing severe
respiratory chain enzyme deficiency in humans and yeast
Emma L. Blakely
1
, Anna L. Mitchell
1
, Nicholas Fisher
2
, Brigitte Meunier
2
, Leo G. Nijtmans
3
,
Andrew M. Schaefer
1
, Margaret J. Jackson
4
, Douglass M. Turnbull
1
and Robert W. Taylor
1
1 Mitochondrial Research Group, School of Neurology, Neurobiology and Psychiatry, The Medical School, University of Newcastle upon
Tyne, UK
2 Wolfson Institute for Biomedical Research, University College London, UK
3 Nijmegen Center for Mitochondrial Disorders, Department of Pediatrics, Radboud University Nijmegen Medical Center, the Netherlands
4 Department of Neurology, Royal Victoria Infirmary, Newcastle upon Tyne, UK
The mitochondrial bc
1
complex (ubiquinol–cytochrome
coxidoreductase or complex III; EC 1.10.2.2) is a
membrane-bound enzyme that catalyses the transfer of
electrons from ubiquinol to cytochrome c, coupling
this process to the translocation of protons across the
inner mitochondrial membrane. In higher eukaryotes,
the enzyme complex is composed of 11 polypeptide
subunits. One subunit, cytochrome b, is encoded
by the mitochondrial genome (mtDNA) [1] whilst the
others are encoded by the nucleus and synthesized on
cytoplasmic ribosomes prior to being imported into
mitochondria and assembled into a functional com-
plex. As a hydrophobic, integral membrane protein
consisting of eight transmembrane helices, cyto-
chrome bis fundamental for the assembly and function
of complex III, and together with cytochrome c
1
and
the iron–sulfur protein (ISP) it forms the catalytic core
of the enzyme.
It has become apparent in recent years that dis-
orders of the mitochondrial respiratory chain due to
Keywords
Mitochondrial DNA (mtDNA); complex III;
complex I; cytochrome b; yeast mutants
Correspondence
R.W. Taylor, Mitochondrial Research Group,
School of Neurology, Neurobiology and
Psychiatry, The Medical School, Framlington
Place, University of Newcastle upon Tyne,
NE2 4HH, UK
Fax: +44 191 2228553
Tel: +44 191 2223685
E-mail: r.w.taylor@ncl.ac.uk
(Received 15 April 2005, accepted 19 May
2005)
doi:10.1111/j.1742-4658.2005.04779.x
Whereas the majority of disease-related mitochondrial DNA mutations
exhibit significant biochemical and clinical heterogeneity, mutations within
the mitochondrially encoded human cytochrome bgene (MTCYB) are
almost exclusively associated with isolated complex III deficiency in muscle
and a clinical presentation involving exercise intolerance. Recent studies
have shown that a small number of MTCYB mutations are associated with
a combined enzyme complex defect involving both complexes I and III, on
account of the fact that an absence of assembled complex III results in a
dramatic loss of complex I, confirming a structural dependence between
these two complexes. We present the biochemical and molecular genetic
studies of a patient with both muscle and brain involvement and a severe
reduction in the activities of both complexes I and III in skeletal muscle
due to a novel mutation in the MTCYB gene that predicts the substitution
(Arg318Pro) of a highly conserved amino acid. Consistent with the dra-
matic biochemical defect, Western blotting and BN-PAGE experiments
demonstrated loss of assembled complex I and III subunits. Biochemical
studies of the equivalent amino-acid substitution (Lys319Pro) in the yeast
enzyme showed a loss of enzyme activity and decrease in the steady-state
level of bc
1
complex in the mutant confirming pathogenicity.
Abbreviations
BN-PAGE, Blue-native PAGE; ISP, iron–sulfur protein; MELAS, mitochondrial encephalopathy, myopathy, lactic acidosis and stroke-like
episodes; MERRF, myoclonic epilepsy and ragged-red fibres; MTCYB, mitochondrial cytochrome bgene; mtDNA, mitochondrial DNA;
MRI, magnetic resonance imaging; Q
o
, quinol oxidation site; Q
i
, quinol reduction site; RFLP, restriction fragment length polymorphism;
RRF, ragged-red fibre.
FEBS Journal 272 (2005) 3583–3592 ª2005 FEBS 3583

pathogenic mutations in mtDNA genes are amongst
the most common inherited human metabolic diseases
[2]. Many pathogenic mtDNA mutations are hetero-
plasmic, affecting a population of mitochondrial
genomes within the cell, and all cause disease by a
common mechanism, the impairment of cellular oxida-
tive phosphorylation [3]. Patients can present with a
wide spectrum of clinical phenotypes affecting predo-
minantly skeletal muscle, heart and the CNS, which is
dependent upon the abundance and segregation of
the causative mutation, the gene that is mutated and
the associated biochemical defect; point mutations in
mitochondrial tRNA genes and large-scale mtDNA
rearrangements cause multiple respiratory chain abnor-
malities through impairment of mitochondrial trans-
lation [4], whilst mutations in protein-coding genes
generally affect only a single enzyme complex [5,6].
A number of patients with respiratory complex III
deficiency due to nonsense, missense or frameshift
mutations in the cytochrome b(MTCYB) gene have
now been described [7]. Although MTCYB mutations
are associated with numerous clinical presentations
including mitochondrial encephalopathy [8,9], cardio-
myopathy [10,11], septo-optic dysplasia [12] and a
multisystem disorder [13], the overwhelming majority
have been reported in patients with severe exercise
intolerance and myopathy, often associated with mus-
cle weakness and ⁄or myoglobinuria and evidence of
mitochondrial proliferation as evidenced by ragged-red
fibres (RRF) on muscle biopsy [14–20]. In many of
these patients the genetic defect arises sporadically,
does not appear to be transmitted to offspring and is
restricted to skeletal muscle with no evidence of the
mutation in mitotic tissues such as peripheral lympho-
cytes. Although this absence of pathogenic mutations
in fibroblasts and platelets precludes their study in
transmitochondrial cybrid cell lines, significant insight
into the underlying molecular mechanism of human
MTCYB mutations has been made by introducing
the equivalent amino-acid substitutions into the yeast
(Saccharomyces cerevisiae) enzyme [21,22].
A curious observation in some patients with
MTCYB mutations is that whilst the genetic defect
resides within a structural component of complex III,
the in vitro assay of respiratory chain enzyme activities
in muscle mitochondria reveals deficiency of both
complexes I (NADH–ubiquinone oxidoreductase) and
complex III [16,18,19]. Conversely, mutation of the
nuclear encoded complex I gene NDUFS4 also results
in a reduction of complex III activity in addition to
complex I deficiency [23]. Recent studies using both
human and mouse cell lines harbouring deleterious
MTCYB mutations have demonstrated a structural
dependence among complexes I and III, highlighting
the need for a fully assembled respiratory complex III
for the stability and activity of complex I [24]. Further-
more, these experiments corroborate previous two-
dimensional blue native electrophoresis data which
revealed that individual respiratory chain complexes
physically interact with each other to form supercom-
plexes [25].
Here we report our studies in a patient with mito-
chondrial encephalopathy and a previously unreported
15 699 GfiCMTCYB mutation that predicts a highly
conserved amino-acid substitution (Arg318Pro) in the
C-terminal portion of the protein. In agreement with
recent findings, analysis of the patient’s muscle biopsy
reveals a severe biochemical deficiency in the activities
of both complex III and complex I which is reflected
by a dramatic loss in the steady state levels of both
complexes, whilst modelling of the equivalent mutation
in yeast (Lys319Pro) reveals a bc
1
defect, thus confirm-
ing the pathogenicity of the human 15 699 GfiC
mtDNA transversion.
Results
Biochemical analysis of patient’s muscle biopsy
reveals a combined deficiency of complexes I
and III
Enzyme histochemistry of the patient’s muscle revealed
normal activities of both cytochrome coxidase (COX)
and succinate dehydrogenase, although approximately
1% of total fibres showed evidence of subsarcolemmal
accumulation of mitochondria, typical of ragged-red
fibres. Measurements of the individual respiratory
chain complexes in patient muscle mitochondria
revealed specific defects involving complexes I and III
activities (Table 1), both of which were decreased to
approximately 5% of control values. Furthermore, the
activities of these enzymes and other respiratory chain
complexes were entirely normal in muscle from both
the patient’s clinically unaffected mother and her sister
(Table 1).
Mitochondrial DNA analysis identifies a novel
MTCYB mutation
The patient’s clinical presentation, together with the
histochemical and biochemical findings in muscle,
prompted us to pursue a mitochondrial genetic abnor-
mality. Following a negative screen for common
pathogenic mtDNA mutations including the 3243
AfiG mutation commonly associated with the
MELAS (mitochondrial myopathy, encephalopathy,
Novel cytochrome bmutation in human and yeast E. L. Blakely et al.
3584 FEBS Journal 272 (2005) 3583–3592 ª2005 FEBS
lactic acidosis and stroke-like episodes) phenotype,
we determined the sequence of the entire coding region
of the mitochondrial genome. This revealed several
common polymorphic sequence variants and a novel
GfiC transversion at position 15 699 in the MTCYB
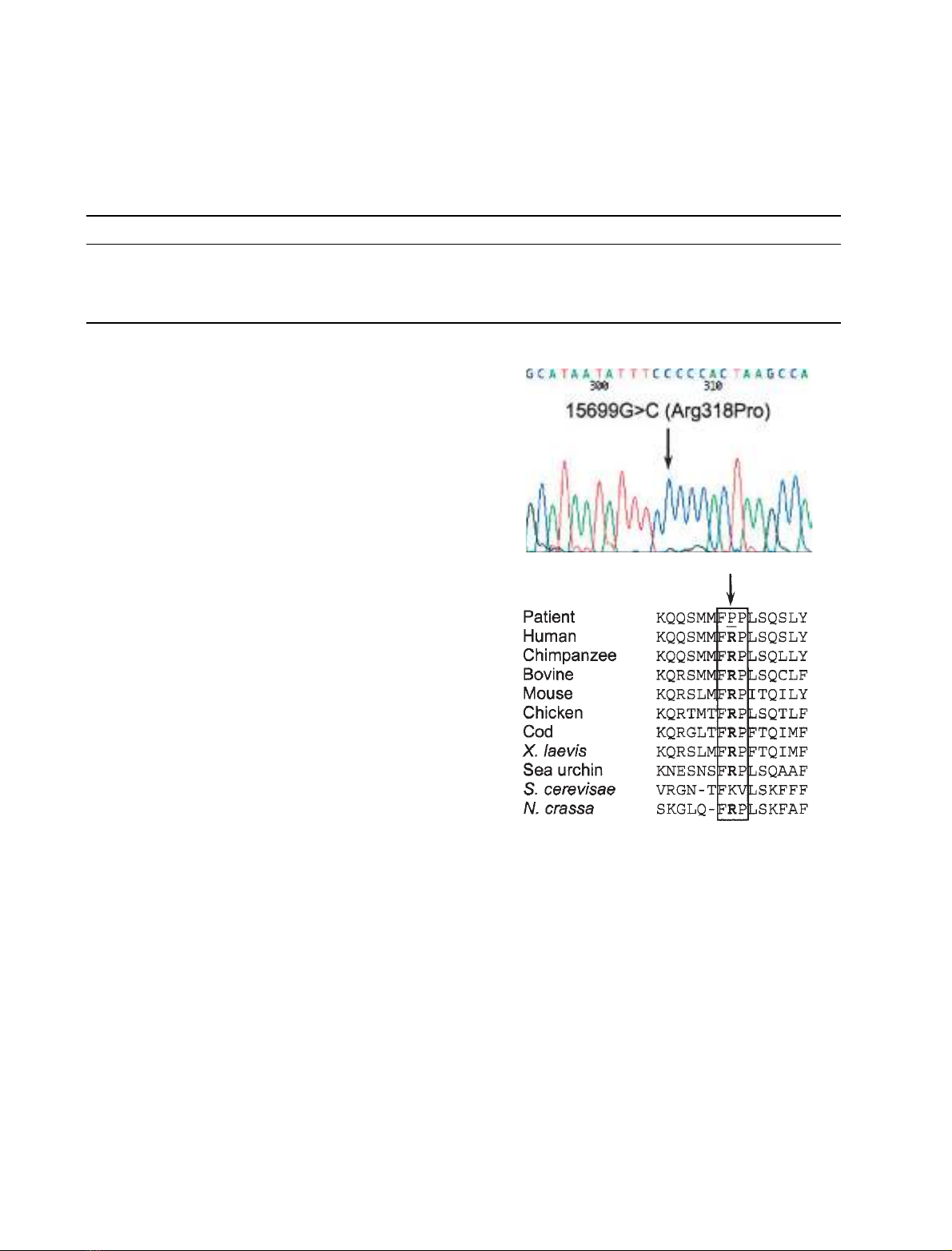
gene (Fig. 1A) that had not previously been reported
[7]. The 15 699 GfiC mutation, which is predicted to
change an evolutionary conserved arginine to proline
at amino-acid position 318 of the human protein
(Fig. 1B), destroys an MwoI restriction site permitting
the assessment of heteroplasmy at this site by PCR-
RFLP analysis. This clearly demonstrated that the
mutation was heteroplasmic, with highest levels present
in the patient’s muscle (88% mutant load). Consistent
with a pathogenic role for this mutation, lower levels
were evident in mitotic tissues including urinary epithe-
lia (16% mutant load), circulating lymphocytes (13%
mutant load) and hair shafts (14% mutant load).
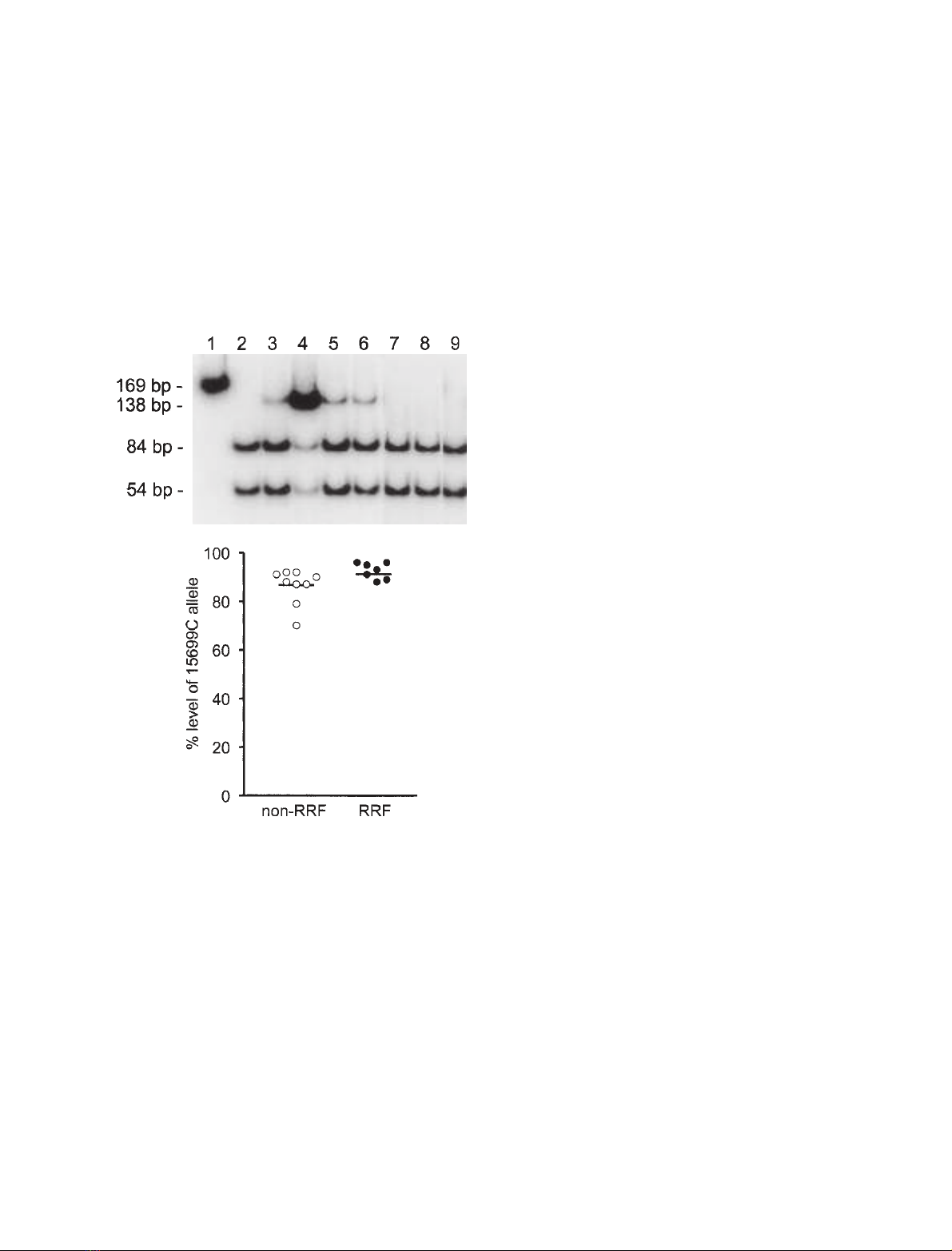
Moreover, the 15 699 GfiC mutation was undetecta-
ble in skeletal muscle from the patient’s mother and
sister and urinary epithelial cells from her son
(Fig. 2A), suggesting that it had arisen de novo in the
patient and had not been transmitted to her child who
was clinically unaffected.
Distribution of 15 699 GfiC mutation
in single fibres
Further supportive evidence to suggest that the 15 699
GfiC mutation was responsible for the muscular
symptoms was provided by studies of individual skel-
etal muscle fibres to determine whether the higher
amounts of mutant mtDNA were present in those
fibres showing mitochondrial accumulation (COX-posi-
tive RRF) than those that did not (non-RRF). Using
PCR-RFLP analysis, we did detect higher levels of
the 15 699 GfiC transversion in RRF [92.6 ± 1.25%
(n¼7)] than non-RRF [86.2 ± 2.43% (n¼9)],
although this just failed to reach statistical significance
(P¼0.0513, Student’s t-test) (Fig. 2B).
The 15 699 GfiC mutation is associated with
decreased steady–state levels of respiratory chain
complex I and III subunits
Western blotting studies were performed to investigate
the effect of the 15 699 GfiC mutation on the expres-
Table 1. Respiratory chain complex activities in skeletal muscle mitochondria. The activities of complexes I and III are markedly decreased
in the patient’s muscle, whilst all respiratory chain complex activities were normal in muscle biopsies from the patient’s mother and sister.
Enzyme activities are expressed as nmol NADH oxidized per min per unit citrate synthase (CS) for complex I, nmol DCPIP reduced per min
per unit citrate synthase for complex II (succinate–ubiquinone-1 reductase) and the apparent first-order rate constant per s per unit citrate
synthase for complexes III and IV (x 10
3
). Control values are shown as mean ± SD. DCPIP, 2,6-dichlorophenol-indophenol; SD, standard devi-
ation.
Complex I ⁄CS Complex II ⁄CS Complex III ⁄CS Complex IV ⁄CS
Patient 0.042 0.369 0.094 0.941
Patient’s mother 0.196 0.350 2.020 0.907
Patient’s sister 0.338 0.413 2.133 0.961
Controls 0.240 ± 0.060 0.320 ± 0.088 2.10 ± 0.75 1.34 ± 0.39
A
B
Fig. 1. Identification of a novel 15 699 GfiCMTCYB mutation.
(A) Sequencing electropherogram showing the 15 699 GfiC trans-
version in the patient (arrow) which is predicted to result in an
arginine to proline change at position 318. (B) Multiple sequence
alignment of the MTCYB gene highlighting the evolutionary conser-
vation of this region of the protein, including the positively charged
arginine at position 318 (shown in bold) and the two flanking amino
acids (boxed). Interestingly, the equivalent residue in yeast
(Lys319) is similarly positively charged.
E. L. Blakely et al.Novel cytochrome bmutation in human and yeast
FEBS Journal 272 (2005) 3583–3592 ª2005 FEBS 3585
sion of mitochondrial respiratory chain subunit pro-
teins. In agreement with enzyme activity measure-
ments, immunoblotting with a panel of monoclonal
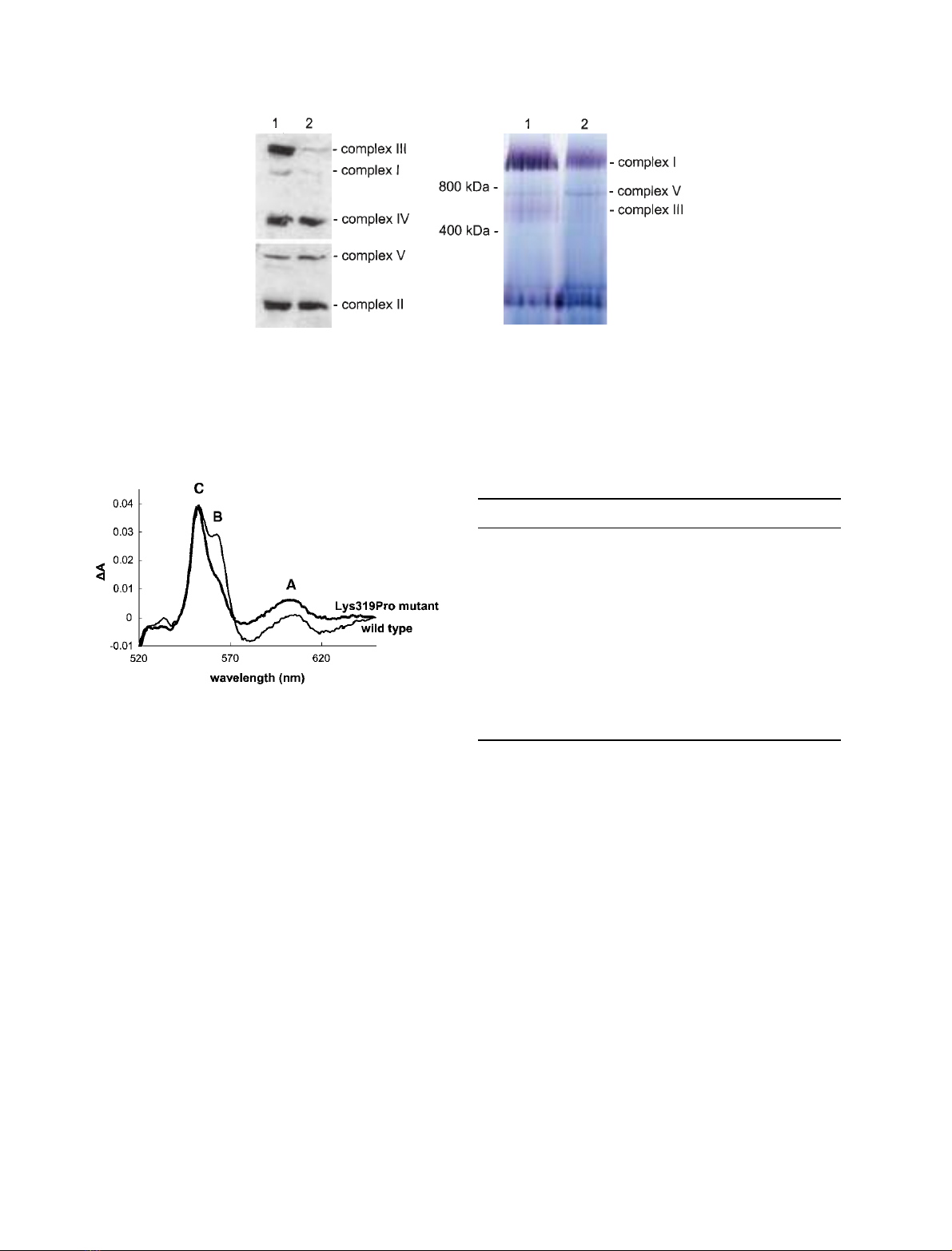
antibodies demonstrated a severe and almost complete
reduction in the amount of complex I (NDUFA9) and
complex III (Core 2 protein) subunits in patient mus-
cle, whereas the steady-state levels of other respiratory
chain subunits were unaffected (Fig. 3A). BN-PAGE
and in-gel activity assays further confirmed the
observed decrease in both complex I and III activities
(Fig. 3B).
Modelling the human Arg318Pro mutation
in yeast
Arg318 is very highly conserved in the vertebrate,
plant and bacterial sequence data (Fig. 1B), but is
replaced by another positively charged residue (lysine)
in yeast, in which the equivalent residue is Lys319. To
investigate further the mechanism by which the 15 699
G > C mutation disrupts complex III activity, we
used biolistic transformation to generate a yeast
mutant harbouring the Lys319Pro mutation. As with
all yeast mitochondrial mutants, the strain was homo-
plasmic with every copy of the mitochondrial genome
containing the mutation. First, we investigated the
effect of the mutation on the level of cytochromes in
whole cells. Cytochrome bcontent (based on the dithio-
nite reduced spectra in the visible region (Fig. 4) was
decreased by 50% of the wild type strain, while the
cytochromes cand oxidase contents were not affected
(Table 2). It seems therefore that the Lys319Pro muta-
tion hinders the assembly of the complex, an obser-
vation which might have been predicted from the
structural analysis. Lys319 is located at the C-terminal
region of a b-turn linking helices F2 and G (Fig. 5)
and probably acts as a backbone H-bond donor to
Asn316. Mutation of Lys319 to a proline would
remove this H-bond and probably be disruptive for
the geometry of the turn. Lys319 approaches with
4.5 A
˚of the 14 kDa subunit (although it is not
involved in strong interactions with this subunit) and
as such, any minor alteration of the architecture of the
associated turn may disrupt this interaction.
Mitochondrial membranes were prepared from the
wild type and mutant strains and the cytochrome c
reductase activity monitored spectrophotometrically as
a function of decylubiquinol (QH
2
) concentration. The
apparent V
m
and K
M
for QH
2
were calculated from
initial rate measurements using derived Eadie–Hofstee
plots (Table 2). The catalytic activity of the mutant
was decreased compared to wild type (V
m
value:
40 s
)1
, 50% of the wild-type rate). The mutation
decreased the K
M
for quinol from 18 to 5 lm. The k
min
(an apparent second-order rate constant equal to
V
m
⁄K
M
) value for Lys319Pro was 8 lm
)1
Æs
)1
(com-
pared to 4.4 lm
)1
Æs
)1
for the wild type). The lower K
M
value indicates that the Q
o
site becomes saturated with
quinol at a lower concentration than the wild type,
suggesting slower rates of electron transfer from Q
o
.It
could be suggested that the mutation had long distance
effect and distorted the architecture of the Q
o
site.
B
A
Fig. 2. Quantitation of the relative amounts of mutant and wild-type
mtDNA in patient tissues by PCR-RFLP analysis. (A) In wild-type
mtDNA, MwoI cuts the amplified 169 bp product into three frag-
ments of 84 bp, 54 bp and 31 bp (band not shown). The 15 699
GfiC mutation abolishes a restriction site for MwoI, yielding frag-
ments of 138 bp and 31 bp and permitting the detection of mtDNA
heteroplasmy at this site. Lane 1, uncut PCR product; lane 2, con-
trol DNA; lane 3, patient’s blood; lane 4, patient’s muscle; lane 5,
patient’s urinary epithelial cells; lane 6, patient’s hair shafts; lane 7,
muscle from patient’s mother; lane 8, muscle from patient’s sister;
lane 9, urinary epithelial cells from patient’s son. (B) Segregation of
the 15 699 GfiC mutation in COX-positive RRF and COX-positive
non-RRF. The mutation load was determined in individual muscle
fibres by PCR-RFLP analysis, and clearly demonstrates higher mean
levels of the mutation (horizontal bars) segregating with fibres that
show mitochondrial accumulation (RRF, black circles) than non-RRF
(open circles).
Novel cytochrome bmutation in human and yeast E. L. Blakely et al.
3586 FEBS Journal 272 (2005) 3583–3592 ª2005 FEBS
However the sensitivity to Q
o
site inhibitors such as
stigmatellin, myxothiazol and azoxystrobin was not
altered (Table 2). Therefore it is unlikely that the
mutation causes major structural change at the Q
o
site,
which would have explained the change in K
M
.
Additionally, it appeared that the overall stability of
mutant bc
1
complex in crude membrane preparations
was not noticeably different from that of the wild type
strain (based on loss of activity over the course of the
day and sensitivity to increased detergent concentra-
tion). This is in contrast to the previously studied
Gly167Glu mutation, located in an extramembranous
helix close to the hinge region of the ISP. The Gly167-
Glu mutation, which was first documented in a patient
with cardiomyopathy [10], affected the stability of the
enzyme, possibly due to an altered binding of the
ISP on the complex [22]. In the Lys319Pro mutant,
although the steady-state level of bc
1
complex in cells
and membrane samples was decreased, the assembled
complex was stable.
Discussion
On account of the clinical and genetic heterogeneity
exhibited by mitochondrial disorders, the investigation
and diagnosis of patients suspected of a respiratory
chain abnormality remain a considerable challenge.
Although some patients present with a well-recognized
clinical phenotype due to a specific mutation (in either
AB
Fig. 3. Western blotting and Blue native gel electrophoresis. (A) Immunoblotting of human skeletal muscle homogenates. Equal amounts
(15 lg) of protein from a control (lane 1) and the patient (lane 2) were subjected to SDS ⁄PAGE and blotted onto a PVDF membrane prior
to incubation with a cocktail of monoclonal antibodies as described in the Experimental procedures. The patient’s muscle homogenate
demonstrated a remarkable loss of complex I and complex III subunits, but normal steady-state levels of complexes II, IV and V subunits.
(B) BN-PAGE and in-gel activity assays were performed exactly as described [35], with 40 lg muscle mitochondrial protein loaded onto the
gel for both a control subject (lane 1) and the patient (lane 2).
Table 2. Properties of the yeast Lys319Pro mutant.
Parameters Wild type Lys319Pro
Cytochrome content (nmolÆg
)1
)
a
Cytochrome b4.10 2.10
Cytochrome c7.60 7.50
Cytochrome oxidase 0.88 0.84
bc
1
activity
b
V
max
(s
)1
)8040
K
M
(lMdecylubiquinol) 18 5
IC
50
(nM)
c
Antimycin 3.0 3.0
Stigmatellin 2.4 2.4
Myxothiazol 2.7 2.7
Azoxystrobin 21.0 17.0
a
Cytochrome content in intact cells estimated from the dithionite-
reduced spectra.
b
Calculated from the decylubiquinol cytochrome c
reductase assay as described in [21].
c
Concentration in inhibitor
required to obtain 50% bc1 activity as measured as above at 40 lM
decylubiquinol.
Fig. 4. Yeast mitochondrial cytochrome content in intact cells Cells
were grown on YPD plates for 24 h, resuspended in 5% ficoll at a
concentration of around 200 mL of cells per ml and optical spectra
of reduced cell suspensions were obtained as described in [21].
Standard line, wild type spectra; bold line, Lys319Pro mutant spec-
tra. C, cytochrome c; B, cytochrome bc
1
; A, cytochrome oxidase.
E. L. Blakely et al.Novel cytochrome bmutation in human and yeast
FEBS Journal 272 (2005) 3583–3592 ª2005 FEBS 3587

























