
Agmatine oxidation by copper amine oxidase
Biosynthesis and biochemical characterization of
N
-amidino-2-hydroxypyrrolidine
Paolo Ascenzi
1,
*, Mauro Fasano
2,
*, Maria Marino
1
, Giorgio Venturini
1
and Rodolfo Federico
1
1
Department of Biology, University ÔRoma TreÕ, Rome, Italy;
2
Department of Structural and Functional Biology,
University of Insubria, Varese, Italy
The product of agmatine oxidation catalyzed by Pisum
sativum L. copper amine oxidase has been identified by
means of one- and two-dimensional
1
H-NMR spectroscopy
to be N-amidino-2-hydroxypyrrolidine. This compound
inhibits competitively rat nitric oxide synthase type I and
type II (NOS-I and NOS-II, respectively) and bovine trypsin
(trypsin) activity, values of K
i
being (1.1 ± 0.1) ·10
)5
M
(at
pH 7.5 and 37.0 °C), (2.1 ± 0.1) ·10
)5
M
(at pH 7.5 and
37.0 °C), and (8.9 ± 0.4) ·10
)5
M
(at pH 6.8 and 21.0 °C),
respectively. Remarkably, the affinity of N-amidino-
2-hydroxypyrrolidine for NOS-I, NOS-II and trypsin is
significantly higher than that observed for agmatine and
clonidine binding. Furthermore, N-amidino-2-hydroxy-
pyrrolidine and agmatine are more efficient than clonidine in
displacing [
3
H]clonidine (¼1.0 ·10
)8
M
) from specific
binding sites in heart rat membranes, values of IC
50
being
(1.3 ± 0.4) ·10
)9
M
and (2.2 ± 0.4) ·10
)8
M
,respec-
tively (at pH 7.4 and 37.0 °C).
Keywords: copper amine oxidase; agmatine; N-amidino-2-
hydroxypyrrolidine; enzyme inhibition; type 1 imidazoline
receptor binding.
Copper amine oxidase has been identified in bacteria, yeasts,
fungi, plants, and animals. This enzyme is a homodimer of
70- to 90-kDa subunits, each containing a single copper ion
and a covalently bound cofactor formed by the post-
translational modification of the catalytic tyrosyl residue
to 2,4,5-trihydroxyphenylalanine quinone (TPQ) [1–4].
Copper amine oxidase catalyzes the oxidative deamination
of biogenic amines, including mono, di, and polyamines,
neurotransmitters such as catecholamines, histamine and
xenobiotic amines, with substrate preferences depending
upon the enzyme source [1–5]. The copper amine oxidase
catalyzed reactions follow the general scheme:
Eox þR-CH2-NH2!Ered þR-CHO ðreaction 1Þ
Ered þO2þH2O!Eox þNH3þH2O2ðreaction 2Þ
where E
ox
represents the enzyme–quinone, R-CH
2
-NH
2
is
the substrate, E
red
is the enzyme–aminoquinol, and R-CHO
is the product aldehyde. Substrate amines interact directly
with TPQ in the reductive part of the process forming a
Schiff base complex (reaction 1). Proton abstraction of the
substrate, catalyzed by an invariant Asp residue, leads to the
release of product aldehyde and leaves the enzyme in the
reduced aminoquinol form (reaction 1) [1–4]. The oxidative
part (reaction 2) leads to reoxidation of the aminoquinol
cofactor with the release of ammonia and hydrogen
peroxide [1–4].
Copper amine oxidase catalyzes also the oxidation of
agmatine [3–5], which has been recognized to be an impor-
tant bioactive molecule, being identified as a novel neuro-
transmitter and modulator of cardiovascular functions via
binding to type 1 imidazoline (I
1
-R) and a-adrenergic
receptors [6,7]. Interestingly, agmatine inhibits nitric oxide
synthase isoforms [8,9] and induces the release of some
peptide hormones [7]. To date, the product(s) of the copper
amine oxidase catalyzed oxidation of agmatine has not been
identified. Moreover, no information is available on the role
played by the product(s) of agmatine metabolism on cell
function(s). Here, the biosynthesis and the biochemical
characterization of N-amidino-2-hydroxypyrrolidine, the
product of agmatine oxidation by Pisum sativum L. copper
amine oxidase, is reported.
MATERIALS AND METHODS
Proteins
P. sativum copper amine oxidase was purified as previously
reported [10]. Rat nitric oxide synthase type I (NOS-I) was
prepared from the rat brain homogenate [11]. Rat nitric
oxide synthase type II (NOS-II) was prepared from the lung
homogenate of rats treated with E. coli lipopolysaccharide
(10 mgÆkg
)1
) [11]. NOS-I and NOS-II containing specimens
were homogenized at pH 7.5 (5.0 ·10
)2
M
Hepes buffer),
5.0 ·10
)4
M
EGTA, 1.0 ·10
)3
M
dithiothreitol, and
0.1 mgÆmL
)1
phenylmethanesulfonyl fluoride [11]. Then,
Correspondence to P. Ascenzi, Dipartimento di Biologia, Universita
`
ÔRoma TreÕ, Viale Guglielmo Marconi 446, I-00146 Rome, Italy.
Fax: + 39 06 55176321, Tel.: + 39 06 55176329,
E-mail: ascenzi@uniroma3.it
Abbreviations:I
1
-R, type 1 imidazoline receptor; MMFF, Merck
Molecular Force Field; NOS-I, rat nitric oxide synthase type I (neu-
ronal constitutive isoform); NOS-II, rat nitric oxide synthase type II
(inducible isoform); TPQ, 2,4,5-trihydroxyphenylalanine quinone;
trypsin, bovine trypsin.
Enzymes: bovine catalase (EC 1.11.1.6); bovine trypsin (EC 3.4.21.4);
Pisum sativum L. copper amine oxidase (EC 1.4.3.6); rat nitric oxide
synthase type I (EC 1.14.13.39); rat nitric oxide synthase type II
(EC 1.14.13.39).
*Note: These authors contributed equally to this work.
(Received 26 July 2001, revised 17 October 2001, accepted 3 December
2001)
Eur. J. Biochem. 269, 884–892 (2002) ÓFEBS 2002
NOS-I and NOS-II containing homogenates were desalted
by chromatography over disposable PD-10 columns packed
withSephadexG-25medium(AmershamPharmaciaBio-
tech, Uppsala, Sweden). Bovine calmodulin, bovine cata-
lase, bovine serum albumin, bovine trypsin (trypsin),
and horseradish peroxidase were purchased from Sigma
Chemical Co (St Louis, MO, USA). Proteins were of
reagent grade and used without further purification.
Chemicals
Agmatine, aminoantipyrine, N-a-benzoyl-
L
-arginine p-nitro-
anilide, clonidine, 3,5-dichloro-2-hydroxybenzenesulfonic
acid, epinephrine, phenylmethanesulfonyl fluoride, and
Escherichia coli lipopolysaccharide (serotype 0127:B8) were
obtained from Sigma Chemical Co. [
3
H]
L
-arginine (specific
activity 2.0 TBqÆmmol
)1
)and[
3
H]clonidine (specific activity
2.6 TBqÆmmol
)1
) were purchased from NEN
TM
Life
Science Products (Boston, MA, USA). Deuterium oxide
(99.8% isotopic enrichment) was obtained from Cortec
(Paris, France). All the other chemicals were from Merck
AG (Darmstadt, Germany). All products were of analytical
or reagent grade and used without further purification.
Animals
Male Sprague–Dawley rats (from Morini, Italy), 4- to
5-month-old, were housed and acclimatized for 1 week under
controlled temperature (20 ± 1 °C), humidity (55 ± 10%),
and light (from 7 a.m. to 7 p.m) conditions. The rats were
anaesthetized with ether in a fume hood, and organs
removed and rapidly chilled in liquid nitrogen (brain
and lung) or in ice-cold medium solution (2.0 ·10
)2
M
NaHCO
3
; heart). Animal experiments were performed accor-
ding to ethical guidelines for the conduct of animal research.
P. sativum
copper amine oxidase assay
Oxidation of agmatine by P. sativum copper amine oxi-
dase was investigated spectrophotometrically by follo-
wing the formation of a pink adduct (e
515nm
¼2.6 ·
10
4
M
)1
Æcm
)1
), as a result of the oxidation of aminoanti-
pyrine and 3,5-dichloro-2-hydroxybenzenesulfonic acid cat-
alyzed by horseradish peroxidase, at pH 7.0 (1.0 ·10
)1
M
phosphate buffer) and 25.0 °C [5,6,10]. In a typical experi-
ment, 20 lL of a buffered P. sativum copper amine
oxidase solution (1.0 ·10
)1
M
phosphate buffer, pH 7.0)
wereaddedtoabufferedsolution(1.0mL;1.0·10
)1
M
phosphate buffer, pH 7.0) containing the substrate (i.e.
agmatine), aminoantipyrine (1.0 ·10
)4
M
), 3,5-dichloro-
2-hydroxybenzenesulfonic acid (1.0 ·10
)3
M
), and horse-
radish peroxidase (1.5 ·10
)6
M
). The initial velocity for the
enzymatic oxidation of agmatine was then measured.
P. sativum copper amine oxidase activity was also
assayed polarographically with a Clark electrode (Hansa-
tech Instruments Ltd, Norfolk, UK) by following the O
2
consumption, at pH 7.0 (1.0 ·10
)1
M
phosphate buffer)
and 25.0 °C [12]. In a typical experiment, 20 lLofa
buffered agmatine solution (1.0 ·10
)1
M
phosphate buffer,
pH 7.0) were added to a buffered solution (1.0 mL;
1.0 ·10
)1
M
phosphate buffer, pH 7.0) containing
P. sativum copper amine oxidase. The initial velocity for
the enzymatic oxidation of agmatine was then measured.
In the enzyme assay, the P. sativum copper amine oxidase
concentration was 5.0 ·10
)9
M
and the agmatine concen-
tration ranged between 5.0 ·10
)5
M
and 5.0 ·10
)3
M
.The
enzyme activity was linear up to 5 min of incubation and
results were expressed as lmol productÆs
)1
Æ(lmol enzyme)
)1
.
Under all the experimental conditions, the initial velocity for
the P. sativum copper amine oxidase catalyzed oxidation of
agmatine was unaffected by the enzyme/substrate incuba-
tion time. In fact, the enzyme/substrate equilibration time
was very short, being completed within the mixing time
(15 s).
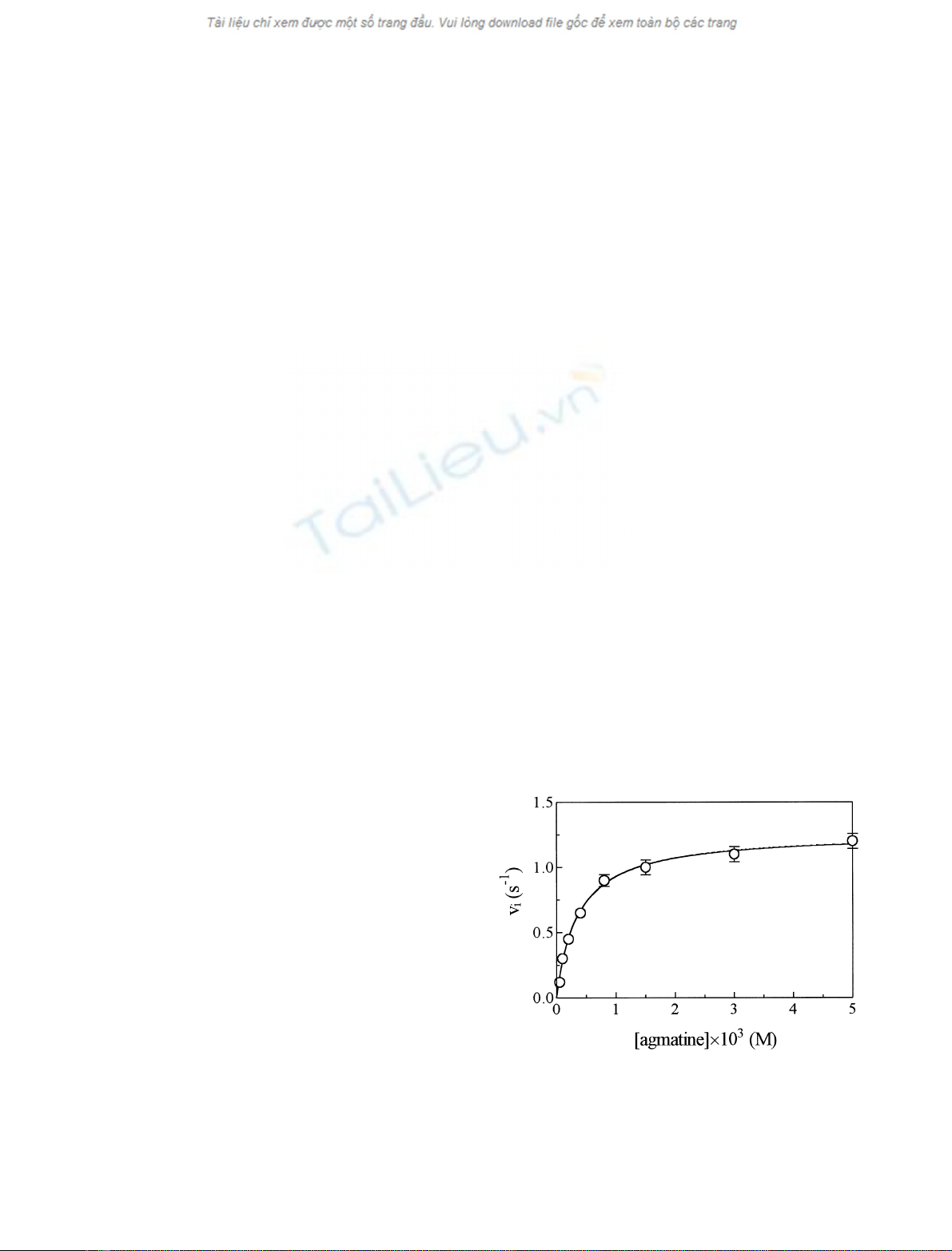
Values of the first-order rate-limiting catalytic constant
(k
cat
) and of the Michaelis constant, as determined in the
absence of the inhibitor (K0
m)fortheP. sativum copper
amine oxidase catalyzed oxidation of agmatine, were
obtained from the dependence of the initial velocity for
agmatine oxidation (v
i
) on the substrate (i.e. agmatine)
concentration ([S]), according to Eqn (1) [13]:
vi¼kcat½S=ðK0
mþ½SÞ ð1Þ
Values of k
cat
and K0
mfor the P. sativum copper amine
oxidase catalyzed oxidation of agmatine are 1.3 ± 0.1 s
)1
and (3.8 ± 0.3) ·10
)4
M
, respectively, at pH 7.0 (1.0 ·
10
)1
M
phosphate buffer) and 25.0 °C (Fig. 1). Values of
k
cat
and K0
mare independent of the enzyme assay.
Biosynthesis of
N
-amidino-2-hydroxypyrrolidine
N-Amidino-2-hydroxypyrrolidine was synthesized as fol-
lows. Twenty micrograms of P. sativum copper amine
oxidase were added to 1.0 mL of a buffered 2.0 ·10
)3
M
agmatine solution (5.0 ·10
)2
M
phosphate buffer, pH 7.4).
28 lg of bovine catalase were also added to the reaction
solution (1.0 mL) in order to remove H
2
O
2
, arising from the
P. sativum copper amine oxidase catalyzed oxidation of
agmatine. The reaction solution was stirred vigorously at
25.0 °C for 20 min, and the product recovered by ultrafil-
tration on Amicon PM10 membranes (Amicon, Inc.,
Beverly, MA, USA).
Fig. 1. Effect of substrate (i.e. agmatine) concentration on values of v
i
for the P. sativum copper amine oxidase catalyzed oxidation of agma-
tine. The continuous line was calculated according to Eqn (1), with the
following values of k
cat
(¼1.3±0.1s
)1
)andK0
m[¼(3.8 ± 0.3) ·
10
)4
M
]. Data were obtained at pH 7.0 and 25.0 °C, mean ± SD. For
further details, see text.
ÓFEBS 2002 N-Amidino-2-hydroxypyrrolidine characterization (Eur. J. Biochem. 269) 885
The total conversion of agmatine to N-amidino-2-
hydroxypyrrolidine was detected by
1
H-NMR spectroscopy.
Moreover, the agmatine/N-amidino-2-hydroxypyrrolidine
stoichiometry is 1 : 1 as shown by
1
H-NMR spectroscopy.
The N-amidino-2-hydroxypyrrolidine concentration was
determined from 100% conversion of agmatine to
N-amidino-2-hydroxypyrrolidine as demonstrated by
1
H-NMR spectroscopy.
Under all the experimental conditions, the formation of
free 4-guanidinobutyraldehyde was observed neither by
the o-aminobenzaldehyde assay [14] (data not shown) nor
1
H-NMR spectroscopy (Figs 2 and 3).
NMR spectroscopy
P. sativum copper amine oxidase catalyzed oxidation of
agmatine was conducted as described above, in deuterated
phosphate buffer (pD 7.4; uncorrected pH-meter reading
7.0); residual oxygen was removed with a mild nitrogen
stream. A control spectrum was recorded prior to addition
of P. sativum copper amine oxidase.
1
H-NMR one- and
two-dimensional spectra were recorded at 25.0 °Cona
Bruker AVANCE 600 NMR spectrometer (Bruker Ana-
lytik, Rheinstetten, Germany), operating at a magnetic field
strength of 14.1 T. The residual water signal was suppressed
by a 2-s presaturation before the observation pulse. The
duration of the pulse corresponding to a flip angle of 90°
was 7.4 ls. The spin system of the agmatine oxidation
product was assigned by COSY, by setting the flip angle of
the second pulse to 35°. To this purpose, 256 t
1
increments
were recorded (4096 points each). The resulting matrix was
zero-filled to 1024 ·4096 complex points and processed
with a 5°-shifted squared sinebell in both dimensions [15].
Building of the
N
-amidino-2-hydroxypyrrolidine structure
Energy minimization of the proposed structure of
N-amidino-2-hydroxypyrrolidine was performed on a
Silicon Graphics Octane workstation (SGI, Mountain
View, CA, USA) by using the program
SPARTAN
(Wave-
function Inc., Irvine, CA, USA).
NOS-I and NOS-II assay
NOS-I and NOS-II activity was assessed by evaluating the
conversion of [
3
H]
L
-arginine to [
3
H]
L
-citrulline at pH 7.5
(5.0 ·10
)2
M
Hepes buffer) and 37.0 °C, in the absence and
presence of N-amidino-2-hydroxypyrrolidine. In a typical
experiment, a NOS-I or NOS-II aliquot (50 lL) was added
to the reaction mixture (100 lL) containing 1.0 ·10
)3
M
NADPH, 1.2 ·10
)3
M
CaCl
2
,1.0lgÆmL
)1
calmodulin,
1.0 ·10
)5
M
FAD, 1.0 ·10
)5
M
FMN, [
3
H]
L
-arginine
(from 12 to 185 kBq) and
L
-arginine (from 1.0 ·10
)6
M
to 1.0 ·10
)4
M
), in the absence and presence of N-ami-
dino-2-hydroxypyrrolidine (from 5.0 ·10
)6
M
and 5.0 ·
10
)5
M
). For the determination of NOS-II activity, CaCl
2
and calmodulin were omitted, and 1.0 ·10
)3
M
EGTA was
added to the reaction mixture. NOS-I and NOS-II activity
was assayed in the presence of 5.0 ·10
)5
M
BH
4
[16]. In the
enzyme assay, the NOS-I or NOS-II concentration was
2.0 ·10
)7
M
. After 15 min incubation, the reaction was
stopped by addition of an ice-cold 2.0 ·10
)2
M
Hepes
buffer solution (700 lL), pH 5.5, containing 2.0 ·10
)3
M
EDTA. [
3
H]
L
-citrulline was separated from [
3
H]
L
-arginine
by ion exchange chromatography on Dowex 50WX8
(Fluka Chemie AG) [11,16]. The enzyme activity was
linear up to 30 min of incubation and results were expressed
as pmol productÆmin
)1
Æ(mg protein)
)1
. Under all the
experimental conditions, the initial velocity for NOS-I and
NOS-II catalyzed conversion of
L
-arginine to
L
-citrulline
was unaffected by the enzyme/inhibitor/substrate incuba-
tion time. In fact, the enzyme/inhibitor/substrate equilibra-
tion time was very short, being completed within the mixing
time (15 s).
Values of the first-order rate-limiting catalytic constant
(k
cat
) and of the Michaelis constant, as determined in the
absence and presence of the inhibitor (K0
mand Kapp
m,
respectively), for NOS-I and NOS-II catalyzed conversion
of
L
-arginine to
L
-citrulline were obtained from the depen-
dence of the initial velocity for substrate conversion (v
i
)on
the
L
-arginine concentration ([S]), according to Eqn (1) [13].
Values of k
cat
and K0
mfor the NOS-I catalyzed conversion
of
L
-arginine to
L
-citrulline were 1.4 ± 10
2
pmol prod-
uctÆmin
)1
Æ(mg protein)
)1
and 4.0 ·10
)6
M
, respectively, at
pH 7.5 and 37.0 °C [11]. Values of k
cat
and K0
mfor the
NOS-II catalyzed conversion of
L
-arginine to
L
-citrulline
were 4.7 ·10
1
pmol productÆmin
)1
Æ(mg protein)
)1
and
1.8 ·10
)5
M
, respectively, at pH 7.5 and 37.0 °C [17].
NO production was also monitored spectrophotometri-
cally (between 350 and 460 nm) following the NO-mediated
conversion of human oxy-hemoglobin (6.0 ·10
)6
M
), added
to the NOS-I and NOS-II preparations, to met-hemoglobin,
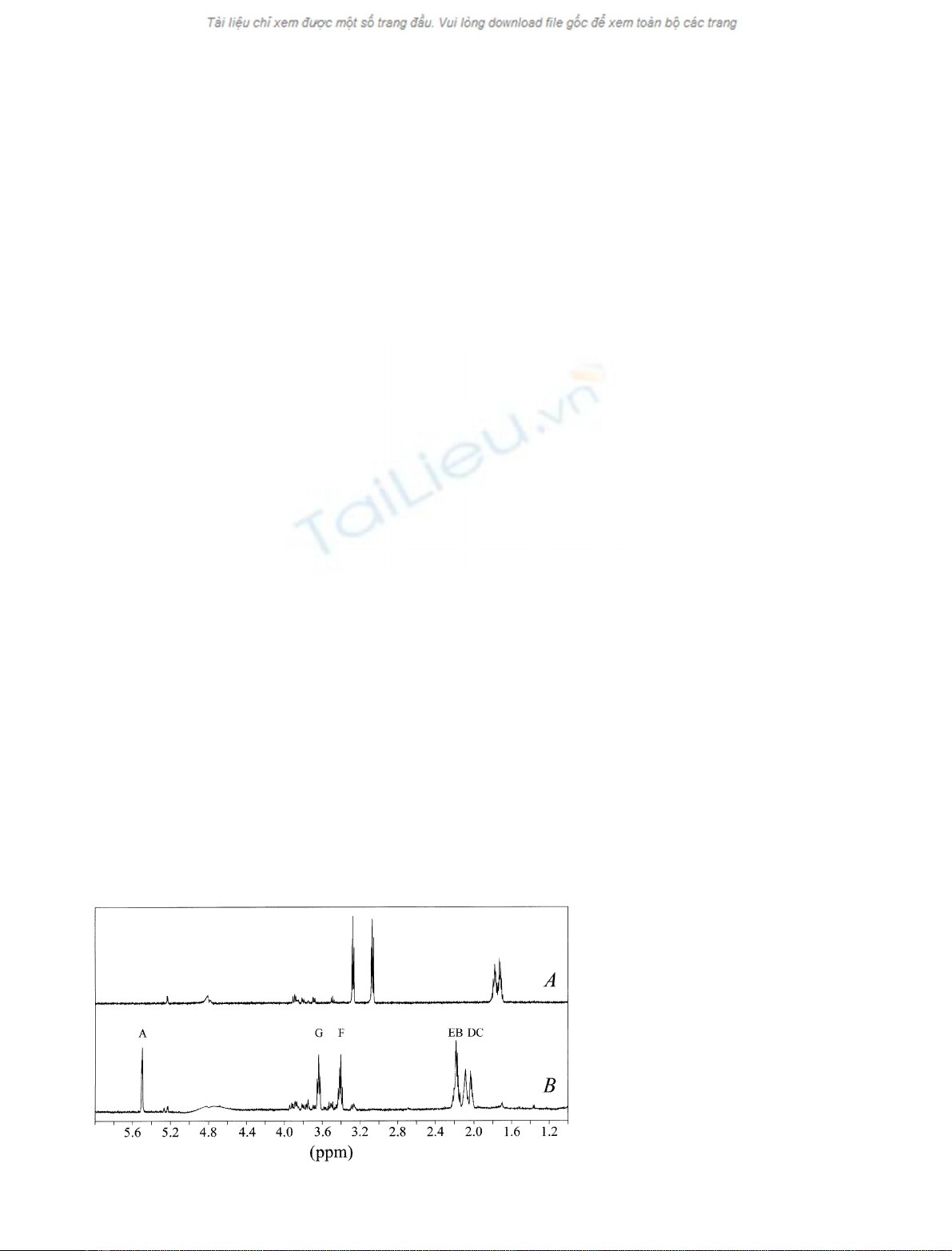
Fig. 2.
1
H-NMR spectra of 2.0 ·10
)3
M
agmatine before (A) and after (B) oxidation
catalyzed by P. sativum copper amine oxidase,
at pD 7.4 and 25.0 °C. Acquisition param-
eters: 4 scans, flip angle 45°, relaxation delay
2 s. The residual water signal was suppressed
by presaturation. For further details, see text.
886 P. Ascenzi et al. (Eur. J. Biochem. 269)ÓFEBS 2002
in the presence of N-amidino-2-hydroxypyrrolidine as the
substrate instead of
L
-arginine, at pH 7.5 (5.0 ·10
)2
M
Hepes buffer) and 37.0 °C [18,19].
Trypsin assay
The trypsin catalyzed hydrolysis of N-a-benzoyl-
L
-arginine
p-nitroanilide was investigated spectrophotometrically (at
408 nm), at pH 6.8 (1.0 ·10
)1
M
phosphate buffer) and
21.0 °C [20], in the absence and presence of N-amidino-
2-hydroxypyrrolidine. In a typical experiment, 20 lLofa
buffered trypsin solution (1.0 ·10
)1
M
phosphate buffer,
pH 6.8) were added to 1.0 mL of a buffered solution
(1.0 ·10
)1
M
phosphate buffer, pH 6.8) containing the
substrate (i.e. N-a-benzoyl-
L
-arginine p-nitroanilide) and
the inhibitor (i.e. N-amidino-2-hydroxypyrrolidine). The
initial velocity for the enzymatic hydrolysis of N-a-benzoyl-
L
-arginine p-nitroanilide was then measured. In the enzyme
assay, the trypsin concentration was 1.0 ·10
)6
M
,the
N-a-benzoyl-
L
-arginine p-nitroanilide concentration ranged
between 1.0 ·10
)5
M
and 1.0 ·10
)3
M
,andtheN-ami-
dino-2-hydroxypyrrolidine concentration ranged between
2.0 ·10
)5
M
and 8.0 ·10
)5
M
. The enzyme activity was
linear up to 10 min of incubation and results were expressed
as lmol productÆs
)1
Æ(lmol enzyme)
)1
.Underalltheexper-
imental conditions, the initial velocity for the trypsin
catalyzed hydrolysis of N-a-benzoyl-
L
-arginine p-nitroani-
lide was unaffected by the enzyme/inhibitor/substrate
incubation time. In fact, the enzyme/inhibitor/substrate
equilibration time was very short, being completed within
the mixing time (15 s).
Values of the first-order rate-limiting catalytic constant
(k
cat
) and of the Michaelis constant determined in the
absence and presence of the inhibitor (K0
mand Kapp
m,
respectively) for the trypsin catalyzed hydrolysis of
N-a-benzoyl-
L
-arginine p-nitroanilide were obtained from
the dependence of the initial velocity for substrate
hydrolysis (v
i
)ontheN-a-benzoyl-
L
-arginine p-nitroani-
lide concentration ([S]), according to Eqn (1) [13]. Values
of k
cat
and K0
mfor the trypsin catalyzed hydrolysis of
N-a-benzoyl-
L
-arginine p-nitroanilide were 0.70 s
)1
and
3.0 ·10
)4
M
, respectively, at pH 6.8 and 21.0 °C[20].
Determination of values of the inhibition
dissociation equilibrium constant (
K
i
)
for
N
-amidino-2-hydroxypyrrolidine binding
to NOS-I, NOS-II, and trypsin
Values of the inhibition dissociation equilibrium constant
(K
i
) for the competitive inhibition of the NOS-I and NOS-II
catalyzed conversion of
L
-arginine to
L
-citrulline (at pH 7.5
and 37.0 °C) and of the trypsin catalyzed hydrolysis of
N-a-benzoyl-
L
-arginine p-nitroanilide (at pH 6.8 and
21.0 °C) by N-amidino-2-hydroxypyrrolidine were deter-
mined from the linear dependence of the Kapp
m/K0
mratio on
the inhibitor concentration (i.e. [I]), according to Eqn (2)
[13]:
Kapp
m=K0
m¼Kÿ1
i½Iþ1ð2Þ
As expected for a simple competitive inhibition system [13],
values of k
cat
for the NOS-I and NOS-II catalyzed
conversion of
L
-arginine to
L
-citrulline and for the trypsin
catalyzed hydrolysis of N-a-benzoyl-
L
-arginine p-nitroani-
lide were unaffected by the inhibitor concentration within
the standard deviation (± 5%).
Model building of the NOS-II: and trypsin:
N
-amidino-2-hydroxypyrrolidine complexes
Molecular models of the human NOS-II: and bovine
trypsin:N-amidino-2-hydroxypyrrolidine complexes were
built using the coordinates of the human NOS-II:S-ethyl-
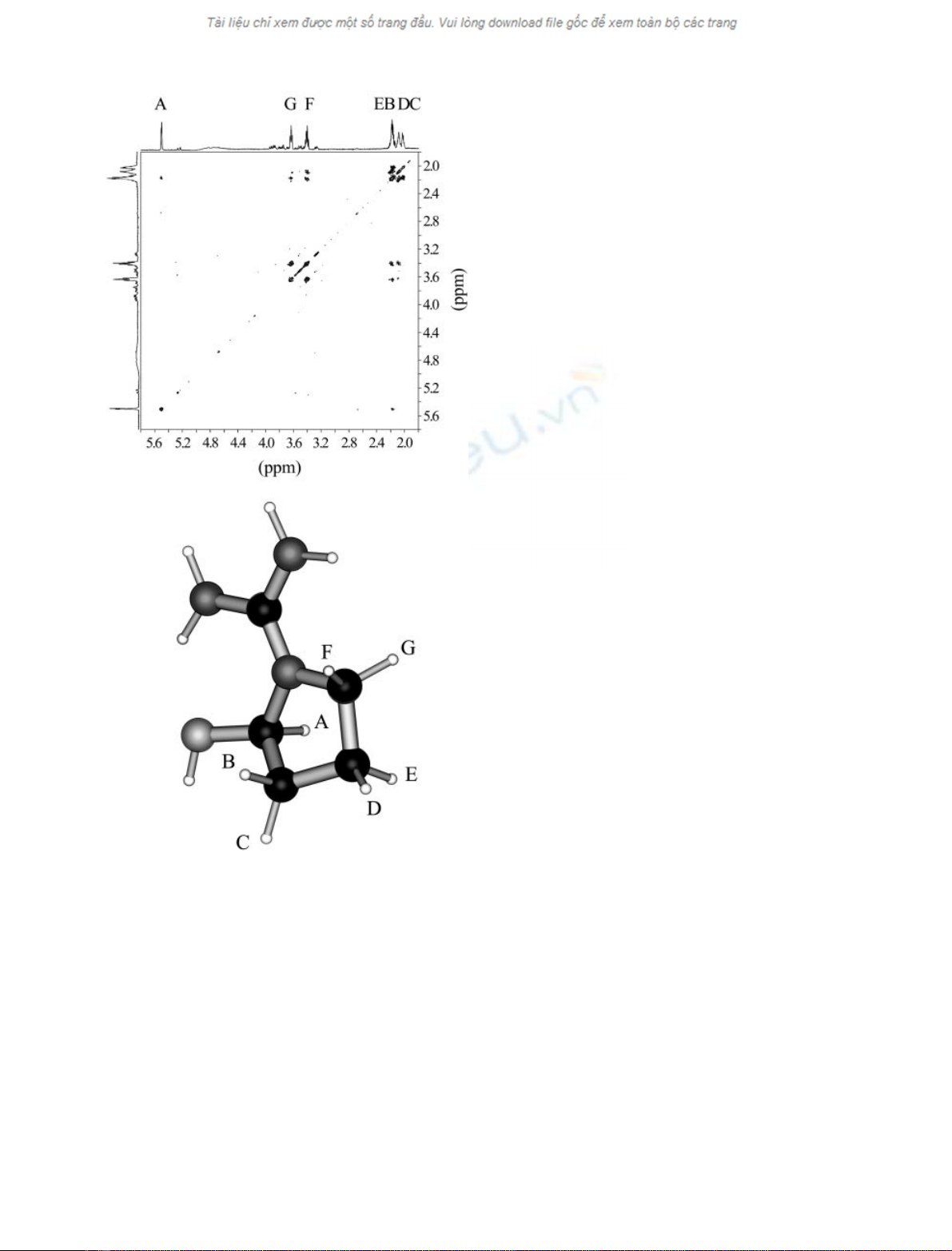
Fig. 3. Two-dimensional COSY spectrum of N-amidino-2-hydroxy-
pyrrolidine, the cyclic oxidation product of agmatine, at pD 7.4 and
25.0 °C(top) and ball-and-stick model of N-amidino-2-hydroxypyrro-
lidine (bottom). Acquisition parameters: 4 scans, 16 dummy scans,
relaxation delay 2 s. Labels refer to the resonance assignment in
Fig. 1B. For further details see text.
ÓFEBS 2002 N-Amidino-2-hydroxypyrrolidine characterization (Eur. J. Biochem. 269) 887

isothiourea complex (PDB accession no. 4NOS) [21] and the
bovine trypsin:benzamidine adduct (PDB accession no.
1CE5) [22] as templates, respectively. The atomic coordi-
nates of rat NOS-II are not yet available [23], the
homologous human enzyme was used instead. The confor-
mations of the N-amidino-2-hydroxypyrrolidine in the
enzyme:inhibitor complexes were obtained after 10 ps
molecular dynamics. Energy minimization and molecular
dynamics were performed on a Silicon Graphics O
2
workstation (SGI, Irvine, CA, USA) with
HYPERCHEM
4.5
for SGI (Hypercube Inc., Gainesville, FL, USA).
I
1
-R binding assay
Cardiac muscle (cleaned of connective tissue and fat) was
finely minced and homogenized in ice-cold medium solution
2.0 ·10
)2
M
NaHCO
3
, containing 1.0 ·10
)4
M
phen-
ylmethanesulfonyl fluoride, with a wet weight to volume
ratio of 1 : 7, using a glass-Teflon homogenizer (10 ·30 s)
[24]. The homogenate was centrifuged at 1500 gfor 15 min
(4.0 °C). The supernatant was centrifuged at 45 000 gfor
5 min (at 4.0 °C). The pellet was washed twice, then
re-suspended in 2 mL of ice-cold 5.0 ·10
)3
M
Hepes buffer,
containing 5.0 ·10
)4
M
EGTA, 5.0 ·10
)4
M
MgCl
2
,and
1.0 ·10
)4
M
ascorbic acid (pH 7.4) [25]. Membrane pre-
parations were free of mitochondria and nuclei as confirmed
by subcellular enzymatic marker assays (data not shown).
Two-hundred and forty micrograms of membrane pro-
tein were incubated for 55 min with 1.3 nmol to 40 nmol
[
3
H]clonidine at 37.0 °C in a final volume of 0.5 mL of
5.0 ·10
)3
M
Hepes buffer, containing 5.0 ·10
)4
M
EGTA,
5.0 ·10
)4
M
MgCl
2
,and1.0·10
)4
M
ascorbic acid
(pH 7.4). The reaction was stopped by rapid vacuum
filtration with a Millipore harvester through Whatman GF/C
glass fiber filters (Whatman International Ltd Maidstone,
UK) presoaked with 10% polyethyleneglycol in Tris/HCl
2.0 ·10
)2
M
, containing MgCl
2
1.0 ·10
)2
M
, followed by
rapid washing of filters with 10 mL ice-cold 5.0 ·10
)3
M
Hepes buffer, containing 5.0 ·10
)4
M
EGTA, 5.0 ·10
)4
M
MgCl
2
,and1.0·10
)4
M
ascorbic acid (pH 7.4). Filters
were placed in a 6-mL scintillation fluid and the radio-
activity determined by liquid scintillation counting. Epine-
phrine (1.0 ·10
)5
M
), which does not bind to imidazoline
sites [26,27], was added to the assay to prevent [
3
H]clonidine
from binding to a-adrenergic receptors. Nonspecific binding
wasdefinedas[
3
H]clonidine-binding (the [
3
H]clonidine
concentration ranged between 1.5 ·10
)4
M
and
5.0 ·10
)4
M
). Saturation studies were performed with
1.0 ·10
)8
M
[
3
H]clonidine and increasing concentrations
of the unlabelled ligand (i.e. N-amidino-2-hydroxypyrroli-
dine, agmatine, and clonidine; from 1.0 ·10
)9
M
to
1.0 ·10
)6
M
). Protein concentration was measured by the
method of Bradford [28], using bovine serum albumin as the
standard.
Values of IC
50
for [
3
H]clonidine displacement from I
1
-R
in heart rat membranes by N-amidino-2-hydroxypyrroli-
dine, agmatine, and clonidine were determined according to
Eqn (3):
a¼1=f1þð½L=IC50Þg ð3Þ
where ais the molar fraction of [
3
H]clonidine bound to I
1
-R
present in heart rat membranes and [L] is the concentration
of the ligand (i.e. N-amidino-2-hydroxypyrrolidine, agma-
tine, or clonidine) [29].
RESULTS
Over the whole substrate (i.e. agmatine) concentration
range explored (i.e. between 5.0 ·10
)5
M
and
5.0 ·10
)3
M
), the P. sativum copper amine oxidase cata-
lyzed oxidation of agmatine follows simple Michaelis–
Menten kinetics (Fig. 1). According to the literature [30],
values of k
cat
and K0
mfor the P. sativum copper amine
oxidase catalyzed oxidation of agmatine are 1.3 ± 0.1 s
)1
and (3.8 ± 0.3) ·10
)4
M
, respectively, at pH 7.0 and
25.0 °C. Moreover, values of k
cat
and K0
mwere independent
of the enzymatic assay used (spectrophotometric vs. pola-
rographic). The stoichiometric analysis of the enzymatic
oxidation of agmatine yields a molar ratio of substrate (i.e.
agmatine) to O
2
and H
2
O
2
of 1 : 1 : 1.
Figure 2 shows the
1
H-NMR spectra of agmatine
before (Fig. 2A) and after (Fig. 2B) oxidation catalyzed
by P. sativum copper amine oxidase, at pD 7.4 and
25.0 °C.Theagmatinesampleshowssomesignalsatthe
impurity level, which however do not hamper the
observation of the main component. The main features
of Fig. 2B with respect to Fig. 2A are: (a) the upset of a
downfield-shifted signal at d¼5.5 p.p.m., and (b) the
splitting of CH
2
signals in magnetically unequivalent
components. On the basis of the general mechanism (see
reactions 1 and 2), one triplet (relative area 1) should
occur at about d¼9 p.p.m., corresponding to the formyl
proton, one triplet at about d¼3 p.p.m. (relative area 2),
and two multiplets at about d¼2 p.p.m. (relative area 2
each). As the -CHO signal was not observed, the
formation of the corresponding free aldehyde (i.e. 4-
guanidobutyraldehyde) was ruled out. To note that the
agmatine/N-amidino-2-hydroxypyrrolidine stoichiometry
is 1 : 1 as shown by
1
H-NMR spectroscopy.
A possible explanation for the resolution of the
magnetic equivalence of CH
2
groups would be the
formation of an intramolecular Schiff base in its emiac-
etalic form, deriving from nucleophilic attack of the
guanidinic
e
N nitrogen to the (transient) aldehydic
carbonyl. This implies the formation of a chiral center
on the ring, with all CH
2
protons consequently becoming
diastereotopic and hence magnetically non equivalent (see
Scheme 1). As the presence of free 4-guanidobutyralde-
hyde was never detected, the formation of the cyclic
product N-amidino-2-hydroxypyrrolidine should occur
within the enzyme catalytic center (shown within square
brackets in Scheme 1).
Figure 3 (top panel) shows the magnitude COSY spec-
trum of the product of agmatine oxidation catalyzed by
P. sativum copper amine oxidase. Starting from the emiac-
etalic proton A, it is possible to walk over the whole spin
system and identify the connectivities on the basis of
3
J
scalar couplings [15]. As three-bond couplings were not
observed, it was assumed that the involved protons form
dihedral angles close to 90°[31]. In other words, the absence
of scalar coupling between A and, say, C identified the axial-
equatorial pairs. Figure 3 (bottom panel) shows the ball-
and-stick model of N-amidino-2-hydroxypyrrolidine (the
product of agmatine oxidation catalyzed by P. sativum
copper amine oxidase) after 200 cycles of energy minimi-
888 P. Ascenzi et al. (Eur. J. Biochem. 269)ÓFEBS 2002

























