
294
NGHIÊN CỨU SỰ HẤP PHỤ MỘT SỐ KIM LOẠI KIỀM THỔ
TRÊN VẬT LIỆU ARMCHAIR SILICENE NANORIBBONS
Trần Minh Tiến1
1. Trường Đại học Thủ Dầu Một
TÓM TẮT
Bài viết trình bày các tính chất của hệ vật liệu armchair silicene nanoribbons (ASiNR) hấp
phụ các nguyên tử kim loại liềm thổ như Be, Mg và Ca. Bằng phương pháp lý thuyết phiếm hàm
mật độ (DFT), dựa trên chương trình mô phỏng lượng tử VASP, một số tính chất đã được khảo
sát như: sự thay đổi về mặt cấu trúc, cấu trúc vng điện tử, mật độ trạng thái, phân bố mật độ
điện tích, sự dịch chuyển điện tích, phân bố mật độ spin đã được tính toán. Kết quả cho thấy cấu
trúc hệ ASiNR có sự thay đổi đáng kể su khi hấp phụ các kim loại kiềm thổ, nhiều nhất là chiều
dài các cạnh của vòng lục giác nằm gần nguyên tử hấp phụ; tối đa lên đến 0.37513 Å đối với
trường hợp hấp phụ Be. Độ mấp mô cấu trúc cũng có thay đổi đáng kể, tối đa lên đến 2.39774 Å
khi hấp phụ Ca. Kết quả cũng chỉ ra rằng có sự xuất hiện bề rộng vng cấm xung quanh mức
Fermi, tối đa đến 0.4744 eV với hệ ASiNR-Mg. Các kết quả cũng chỉ ra có sự dịch chuyển điện
tích tương đối lớn từ vng này đến vùng khác, cung quanh vòng lục giác gần nguyên tử hấp phụ.
Các kết quả cũng cho thấy khả năng tồn tại momen từ ở các cấu trúc sau hấp phụ.
Từ khoá: ASiNR, kim loại kiềm thổ, hấp phụ nguyên tử, vasp
1. GIỚI THIỆU
Nghiên cứu về các vật liệu nano đơn lớp như silicene, phosphorene hoặc germanene đã
rất hấp dẫn trên toàn thế giới. Kể từ khi graphene đơn lớp được phát hiện và chế tạo thành công
vào năm 2004 (K. S. Novoselov và nnk., 2004). Một số cấu trúc 1D-nanoribbons cũng đã được
quan tâm nghiên cứu (Son Y W và nnk, 2006; Li X L và nnk., 2008 ). Đặc điểm cấu trúc, tính
chất điện tử, từ tính, quang học,... của graphene đã được tính toán (Chung H C và nnk, 2008;
Yang L, và nnk, 2007; Lin M F, và nnk, 2000; Li Z, và nnk., 2008; Basu D, và nnk, 2008. Sau
graphene, nhiều hệ thống vật liệu đơn lớp dựa trên các nguyên tử khác cũng được tập trung vào
nghiên cứu như silicene, phosphorene hoặc germanene. Silicene là một cấu trúc, được tạo ra
bởi các nguyên tử silicon, có cấu trúc tổ ong hình lục giác như graphene, nhưng không phẳng
như graphene, có độ mấp mô cấu trúc nhất định. Nhiều nghiên cứu đã tìm cách mở rộng bề rộng
vùng cấm của silicene để tăng khả năng ứng dụng của nó trong các bóng bán dẫn hiệu ứng
trường hiệu suất cao (FET). Quhe Ruge và nnk, 2012 đã nghiên cứu những thay đổi về độ rộng
dải của silicene khi hấp phụ nguyên tử kiềm. Kết quả chỉ ra rằng chiều rộng của vùng cấm có
thể được mở rộng tối đa 0,5 eV. Sahin và nnk, 2013 nghiên cứu về sự hấp phụ của các nguyên
tử kim loại chuyển tiếp kiềm, kiềm thổ và 3D trên silicene. Kết quả cho thấy sự hấp phụ của
các nguyên tử kim loại kiềm vào các vị trí hollow sẽ không gây ra bất kỳ biến dạng cấu trúc
nào. Sivek Jozef và nnk, 2013, công bố kết quả nghiên cứu về sự hấp thụ Bo, N, Al và P trên
silicene. Kết quả chỉ ra rằng các vị trí hấp phụ tối ưu là valley, brige, vallley và các vị trí top
cho các nguyên tử B, N, Al và P, tương ứng. Các hệ cấu trúc đều thể hiện tính kim loại sau khi
hấp phụ, cùng với sự dịch chuyển electron mạnh từ silicene sang nguyên tử B, N và P. Kaloni

295
và nnk, 2014 đã trình bày kết quả nghiên cứu ảnh hưởng của sự hấp phụ kim loại (Au, Hg, Tl và
Pb) đối với silicene. Kết quả cho thấy vị trí hấp phụ tối ưu là các hollow. Nghiên cứu về sự hấp
phụ và phân tán lithium trên silicene với ranh giới hạt (GB) cũng được thực hiện bởi nhóm tác
giả Wang Xiao và nnk, 2019. Kết quả cho thấy sự dịch chuyển của electron Li 2s sang GB làm
tăng đáng kể năng lượng hấp phụ Li, trong khi các rào cản năng lượng nhỏ tạo điều kiện thuận
lợi cho sự di chuyển Li trên bề mặt silicene. Tác giả Phạm Trọng Lâm và nnk, 2020, đã nghiên
cứu Hấp phụ Acetone và Toluene trên Silicene vacancy . Silicene vacancy là một loại silicene
có chứa một nguyên tử silicon bị thiếu duy nhất trong cấu trúc mạng lục giác của nó. Kết quả
chỉ ra rằng năng lượng hấp phụ cho hấp phụ acetone và toluene là -0,36 eV và -0,57 eV, điện
tích dịch chuyển lần lượt là 0,17e và 0,30e. Nghiên cứu về hệ thống cấu trúc 1D cũng rất được
quan tâm. Nhóm tác giả Zhang Jian-Min và nnk, 2014 đã thực hiện một nghiên cứu về pha tạp
nguyên tử P trên ghế bành và ruy băng nano silicene ngoằn ngoèo. Các tính chất cấu trúc, điện
tử và từ tính của hệ thống sau pha tạp đã được các tác giả nghiên cứu. Kết quả cho thấy ASiNR
và ZSiNR là chất bán dẫn không từ tính, ASiNR có pha tạp P ở vị trí cạnh thay đổi thành chất
bán dẫn sắt từ. Tác giả Xu Long và nnk, 2015, đã nghiên cứu các tính chất điện, từ tính và nhiệt
của sự hấp phụ của các nguyên tử Ti trên các dải nano silicene zigzag. Kết quả chỉ ra rằng nguyên
tử Ti, các vị trí ưa thích bên trong các dải ruy băng nano so với trên các cạnh. Hai nguyên tử lân
cận liên kết không liên tục và được hấp phụ ưu tiên ở cùng một phía. Mehdi Aghaei và nnk,
2016 đã nghiên cứu tính ổn định cấu trúc của ruy băng nano silicene chức năng với các cạnh
bình thường, tái tạo và lai. Kết quả cho thấy SiNRs cạnh Klein được fluoride hóa hoàn toàn; đối
với mỗi nguyên tử Si, cạnh được gắn vào ba nguyên tử flo, đây là cấu trúc ổn định nhất.
Trong nghiên cứu này, cấu trúc ASiNR một chiều hấp phụ lần lượt các nguyên tử kim
loại kiềm thổ như Be (ASiNR-Be), Mg (ASiNR-Mg), và Ca (ASiNR-Ca) được nghiên cứu.
2. MÔ HÌNH VÀ PHƯƠNG PHÁP NGHIÊN CỨU
Cấu trúc ô cơ sở của một ASiNR nguyên sơ gồm 28 nguyên tử silicon; các cạnh được
chức hóa bởi 8 nguyên tử hidro để tăng độ bền vững cho cấu trúc. Các nguyên tử alkaline-earth
được đặt ở vị trí hollow, là vị trí tối ưu cho hấp phụ. Các tính toán được thực hiện trên cơ sở lý
thuyết hàm mật độ (DFT), thông qua gói mô phỏng Vienna ab initio (VASP) (G. Kresse và nnk,
1996; G. Kresse và nnk, 1996). Năng lượng trao đổi electron và tương quan được tính toán dựa
trên dạng xấp xỉ gradient tổng quát (GGA) (J.P. Perdew và nnk, 1996), với hàm PBE (Perdew–
Burke–Ernzerhof) – PAW (Projector-Augmented Wave) (G. Kresse, D và nnk, 1999) được sử
dụng. Chức năng sóng và năng lượng trạng thái được xây dựng từ một cơ sở sóng phẳng với
mức cắt năng lượng tối đa là 500 eV. Hướng tuần hoàn của các cấu trúc được xây dựng theo
hướng Oz. Khoảng cách chân không dọc theo hướng giới hạn lượng tử của x và y được đặt lớn
hơn 20 Å để loại bỏ sự tương tác giữa hai dãy lân cận. Các lưới điểm k trong lược đồ Monkhorst-
Pack được sử dụng là 1x2x4 để tính toán tối ưu hóa cấu trúc và 1x2x100 cho các tính toán tự
động tổng hợp. Hệ thống được nới lỏng cho đến khi Lực Hellmann-Feynman nhỏ hơn 0,01
eVÅ1. Sự hội tụ năng lượng được đặt ở 10-5 eV giữa hai bước ion gần nhất.
3. KẾT QUẢ VÀ THẢO LUẬN
3.1. Sự thay đổi cấu trúc sau hấp phụ
Các hệ cấu trúc trước và sau khi hấp phụ được trình bày ở hình 1:
(a)(b)(c)-pristine; (d)(e)(f)-Be;
(g)(h)(k)-Mg; (l)(m)(n)-Ca;
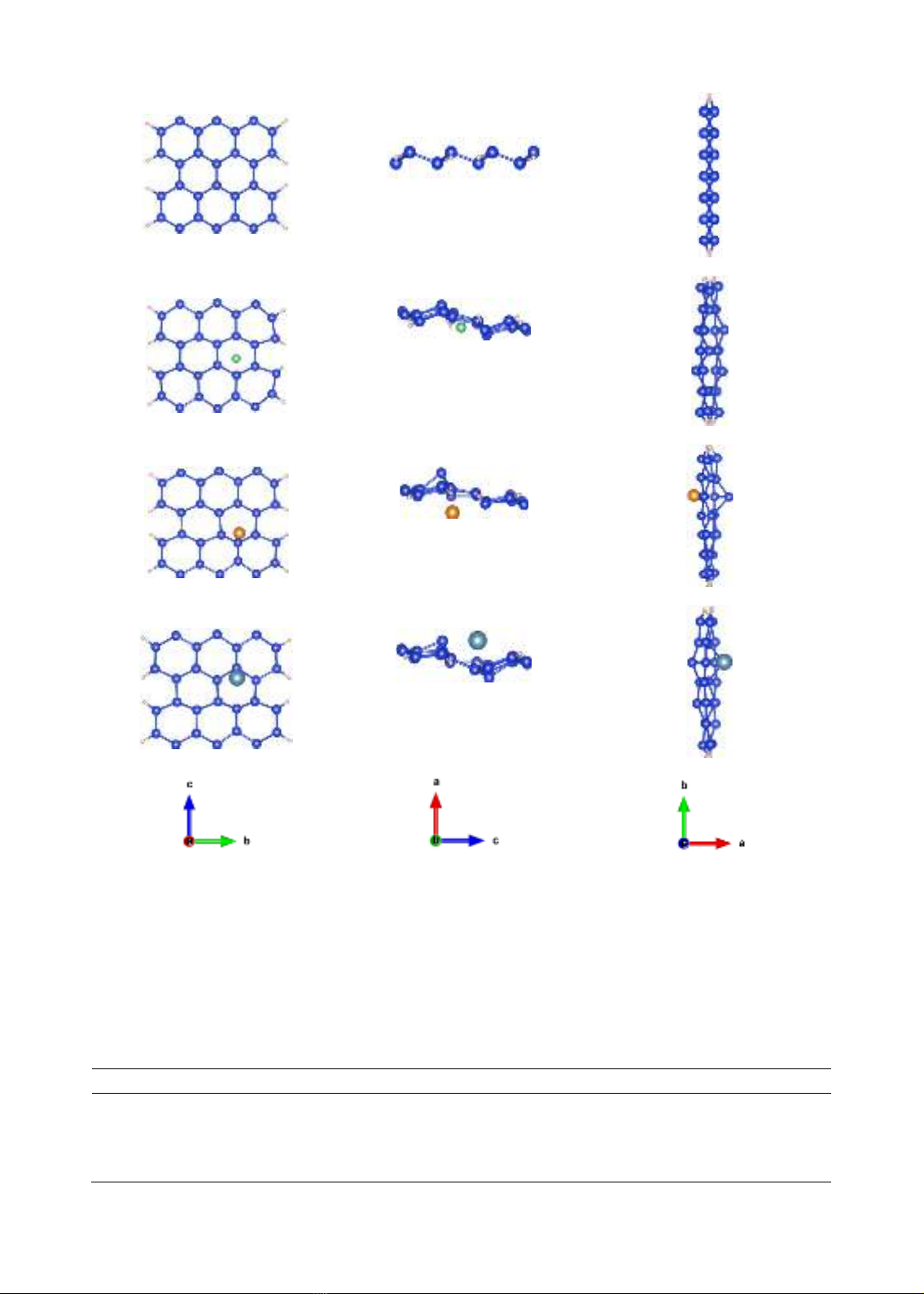
296
(a)
(b)
(c)
(d)
(e)
(f)
(g)
(h)
(k)
(l)
(m)
(n)
Hình 1. cấu trúc trước và sau khi hấp phụ
Cấu trúc sau hấp phụ có nhiều sự thay đổi, đặc biệt là ở vòng lục giác xung quanh nguyên
tử hấp phụ. Sự thay đổi chiều dài của các cạnh và độ mấp mô được trình bày ở bảng 1. Từ một
nguyên tử thuộc vòng lục giác gần vị trí hấp phụ, khoảng cách từ nguyên tử này đến nguyên tử
kế tiếp của vòng lục giác được đặt là khoảng cách thứ nhất, đến nguyên tử thứ 2, 3 được đặt là
các khoảng cách thứ hai và thứ ba. Độ mấp mô cấu trúc được xác định là khoảng cách giữa
nguyên tử cao nhất và thấp nhất theo chiều x.
Bảng 1. Sự thay đổi chiều dài của các cạnh và độ mấp mô
Cấu trúc
1st (Å)
d1 (Å)
2nd (Å)
d2 (Å)
3rd (Å)
d3 (Å)
b(Å)
b (Å)
ASiNR
2.27497
-
3.70681
-
4.15144
-
0.58787
-
ASiNR-Be
2.31239
0.03742
3.85042
0.03742
4.52657
0.37513
2.52959
1.94172
ASiNR-Mg
2.27956
0.00459
3.84735
0.00459
4.38902
0.23758
2.73713
2.14926
ASiNR-Ca
2.33024
0.05527
3.76385
0.05527
4.26278
0.11134
2.98561
2.39774
297
Kết quả cho thấy khoảng cách thứ nhất thay đổi rất ít khi hấp phụ Mg, với Be và Ca thì
lớn hơn, nhưng cũng chỉ vào khoảng từ 0.03 Å đến 0.05 Å. Điều này tương tự đối với các
khoảng cách thứ 2. Thay đổi nhiều nhất là ở các khoảng cách thứ 3; lớn nhất là 0.37513 Å đối
với trường hợp hấp phụ Be, kế tiếp là Mg và Ca với lần lượt 0.23758 Å và 0.11134 Å. Độ mấp
mô cấu trúc cũng thay đổi đáng kể. Độ mấp mô cấu trúc lớn nhất khi hấp phụ Ca, với sự tăng
thêm 2.39774 Å.
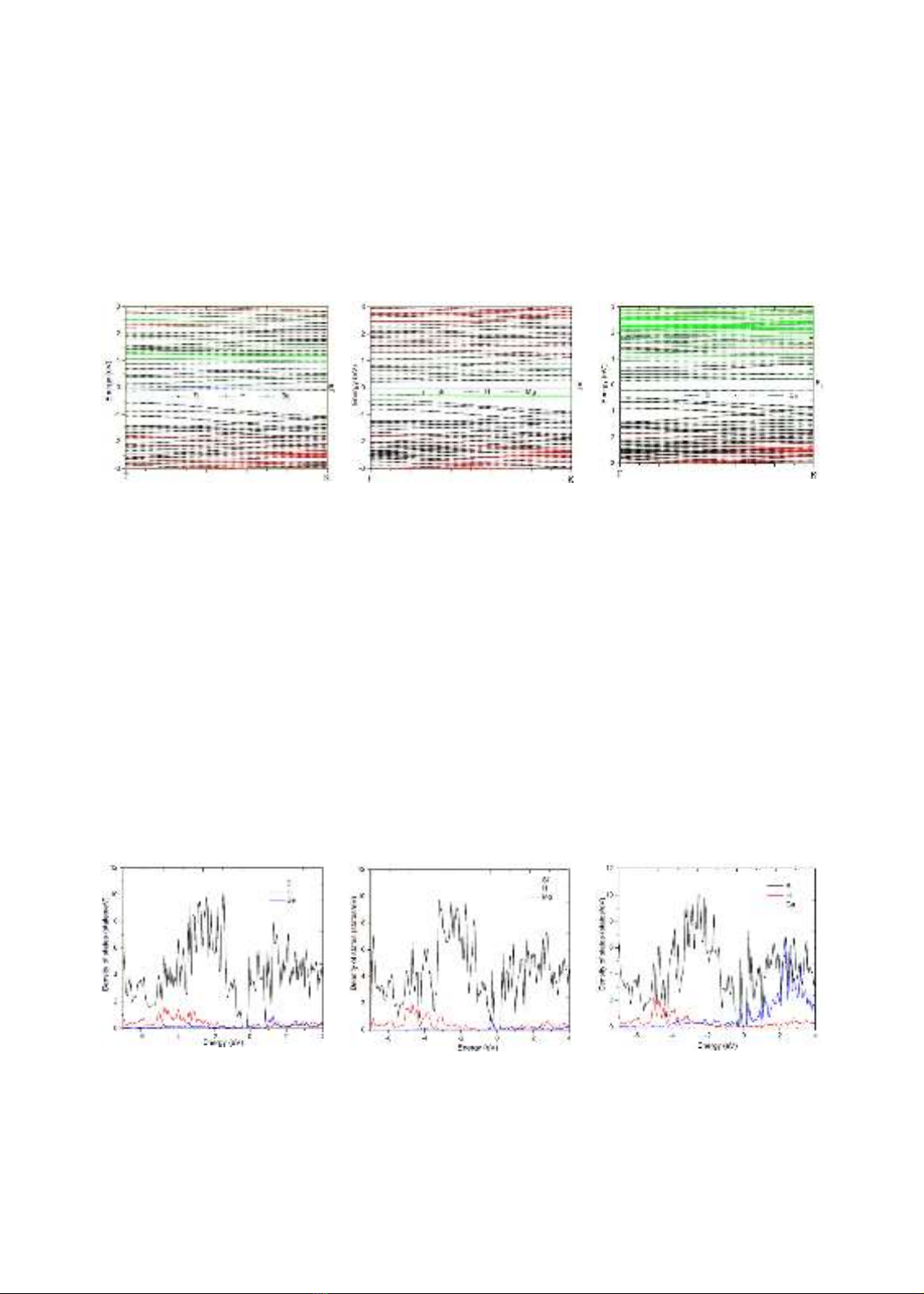
3.2. Cấu trúc vng điện tử
Cấu trúc vùng điện tử của các hệ sau hấp phụ được trình bày trên hình 2.
a) ASiNR-Be
b) ASiNR-Mg
c) ASiNR-Ca
Hình 2. Cấu trúc vùng điện tử
Kết quả cho thấy độ rộng vùng cấm của hệ ASiNR-Be gần như không đáng kể, chỉ vào
khoảng 0.0902 eV, độ rộng vùng cấm lớn nhất đối với hệ ASiNR-Mg là 0.4744 eV, đối với
ASiNR-Ca là 0.3689 eV. Mức độ đóng góp của các nguyên tử rất khác nhau. Nguyên tử Si
đóng góp chủ yếu ở tất cả các cấu trúc, và đóng góp mạnh mẽ nhất ở vùng hóa trị sâu. Các
nguyên tử hấp phụ đóng góp khác nhau vào cấu trúc vùng cho riêng từng cấu hình. Với hệ
ASiNR-Be, nguyên tử Be chủ yếu đóng góp nhẹ vào vùng dẫn, gần mức Fermi. Trong khi
nguyên tử Mg đóng góp vào vùng hóa trị, nằm gần mức Fermi cho cấu trúc ASiNR-Mg. Nguyên
tử Ca đóng góp mạnh mẽ vào vùng dẫn, cách xa mức Fermi hơn so với các trường hợp hấp phụ
Be, Mg. Các kết quả chỉ ra rằng hệ ASiNR hấp phụ các nguyên tử alkaline-earth metals thể hiện
tính bán dẫn, rõ ràng nhất với trường hợp hấp phụ calcium.
3.3. Mật độ trạng thái điện tử
Mật độ trạng thái điện tử sẽ cho đánh giá rõ hơn tính chất cấu trúc vùng điện tử. Mật độ
trạng thái điện tử của các hệ được trình bày ở hình 3.
a) ASiNR-Be
b) ASiNR-Mg
c) ASiNR-Ca
Hình 3. Mật độ trạng thái điện tử
Kết quả cho thấy rõ đóng góp chủ yếu của Si ở cả ba cấu hình, thể hiện qua đường biểu
diễn cao vượt trội hơn so với H và của các nguyên tử hấp phụ. Đóng góp nhiều nhất ở vùng hóa
trị, xung quanh mức năng lượng từ -3 eV đến -2 eV. Xung quang mức Fermi tồn tại vùng không
298
mật độ trạng thái điện tử, cho thấy sự tồn tại của band gap như nhận định từ cấu trúc vùng.
Trong ba trường hợp hấp phụ, chỉ có Ca đóng góp đáng kể ảnh hưởng đến cấu trúc vùng điện
tử, tập trung ở vùng dẫn, xung quanh mức năng lượng 2 eV đến 3 eV. Đây cũng là cấu trúc tiềm
năng nhất cho ứng dụng vào lĩnh vực điện tử nano.
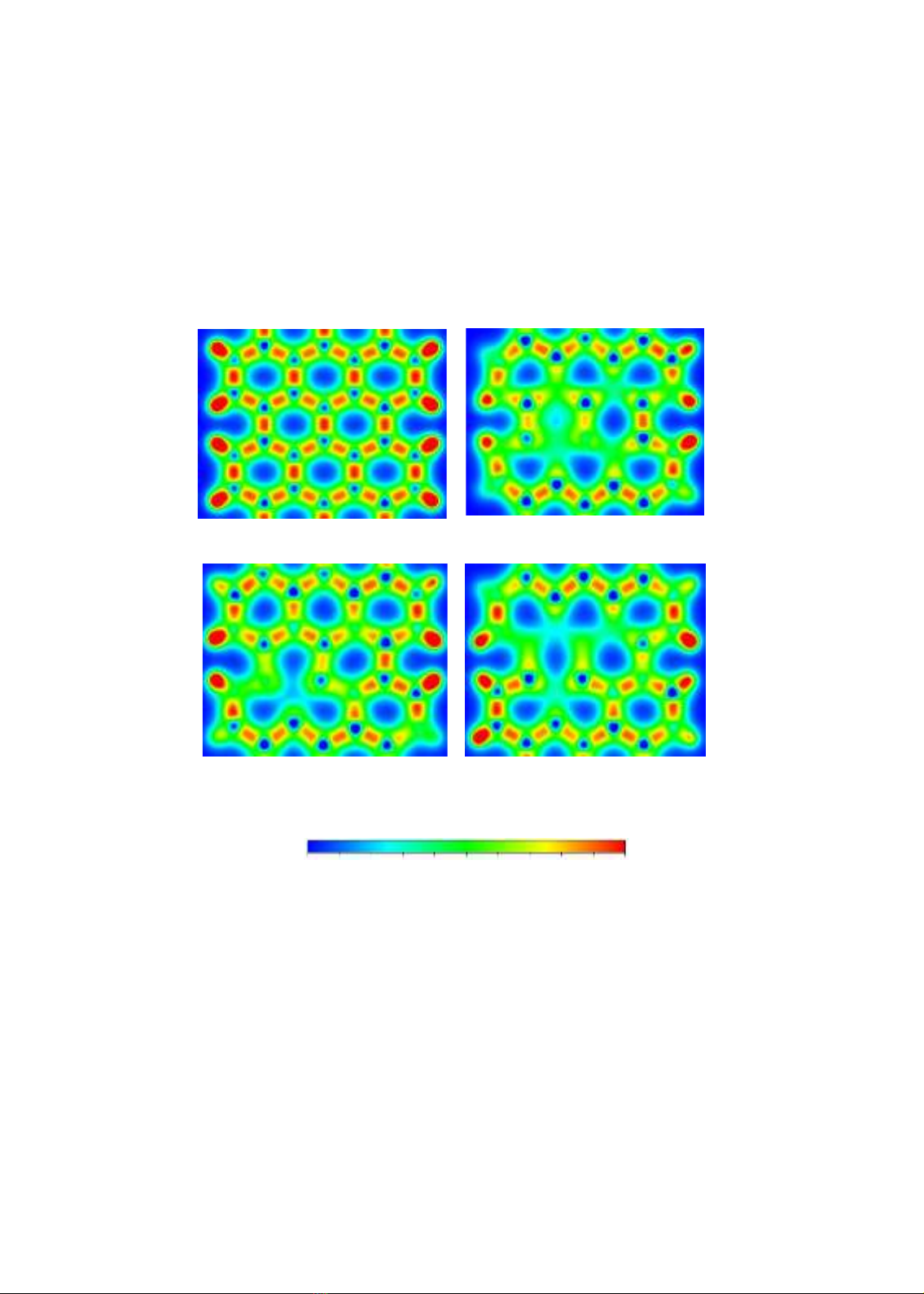
3.4. Phân bố mật độ điện tích
Phân bố mật độ điện tích của các hệ ASiNR-pristine và sau hấp phụ các kim loại kiềm
thổ được trình bày ở hình 4. Ở hình 4a thể hiện phân bố mật độ điện tích của cấu hình ASiNR
nguyên sơ; các hình 4b, 4c, 4d lần lượt trình bày phân bố mật độ điện tích của các cấu hình sau
hấp phụ các nguyên tử Be, Mg, Ca.
a) ASiNR-pristine
b) ASiNR-Be
c) ASiNR-Mg
d) ASiNR-Mg
Hình 4. Phân bố mật độ điện tích
Kết quả cho thấy có sự dịch chuyển điện tích mạnh mẽ ở các hệ sau hấp phụ so với cấu
trúc pristine ban đầu. Thay đổi nhiều nhất ở vị trí vòng lục giác xung quanh nguyên tử hấp phụ.
Tại đây, điện tự có xu hướng bị dịch chuyển về, tạo ra vùng âm hơn so với ban đầu, và các vùng
khác. Sự dịch chuyển điện tích mạnh mẽ nhất khi hấp phụ nguyên tử Ca, vùng dương xung
quanh các liên kết giữa hai nguyên tử Si ban đầu hầu như mất đi.
3.5. Sự dịch chuyển điện tích
Sự khác biệt mật độ điện tích giữa các cấu trúc trước và sau hấp phụ được trình bày ở
hình 5. Các hình 5a, 5b, 5c lần lượt chỉ ra sự dịch chuyển điện tích khi cấu trúc ASiNR hấp phụ
các nguyên tử Be, Mg và Ca.
0
0.1
e/a03










![Bài tập Vật lý sóng: Tổng hợp bài tập 6 [kèm lời giải chi tiết]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20250805/oursky04/135x160/401768817575.jpg)














