
Two 1 : 1 binding modes for distamycin in the minor groove
of d(GGCCAATTGG)
Koen Uytterhoeven
1
, Jiri Sponer
2
and Luc Van Meervelt
1
1
Biomolecular Architecture, Department of Chemistry, Katholieke Universiteit Leuven, Belgium;
2
Institute of Biophysics,
Academy of Sciences of the Czech Republic, and National Center for Biomolecular Research, Brno, Czech Republic
Single-crystal X-ray structure determinations of the complex
between the minor-groove binder distamycin and
d(GGCCAATTGG) reveal two 1 : 1 binding modes which
differ in the orientation of the drug molecule in the minor
groove. The two crystals were grown from different cry-
stallization conditions and found to diffract to 2.38 and
1.85 A
˚, respectively. The structures were refined to comple-
tion using SHELXL-93, resulting in a residual Rfactor of
20.30% for the 2.38-A
˚resolution structure (including 46
water molecules) and 19.74% for the 1.85-A
˚resolution
structure (including 74 water molecules). In both orienta-
tions, bifurcated hydrogen bonds are formed between the
amide nitrogen atoms of the drug and AT base pairs. With a
binding site of at least five base pairs, close contacts between
the terminal distamycin atoms and guanine amino groups
are inevitable. The detailed nature of several of these inter-
actions was further investigated by ab initio quantum
chemical methods.
Keywords: distamycin; drug–DNA complex; minor groove
binder; quantum chemical calculations; X-ray structure.
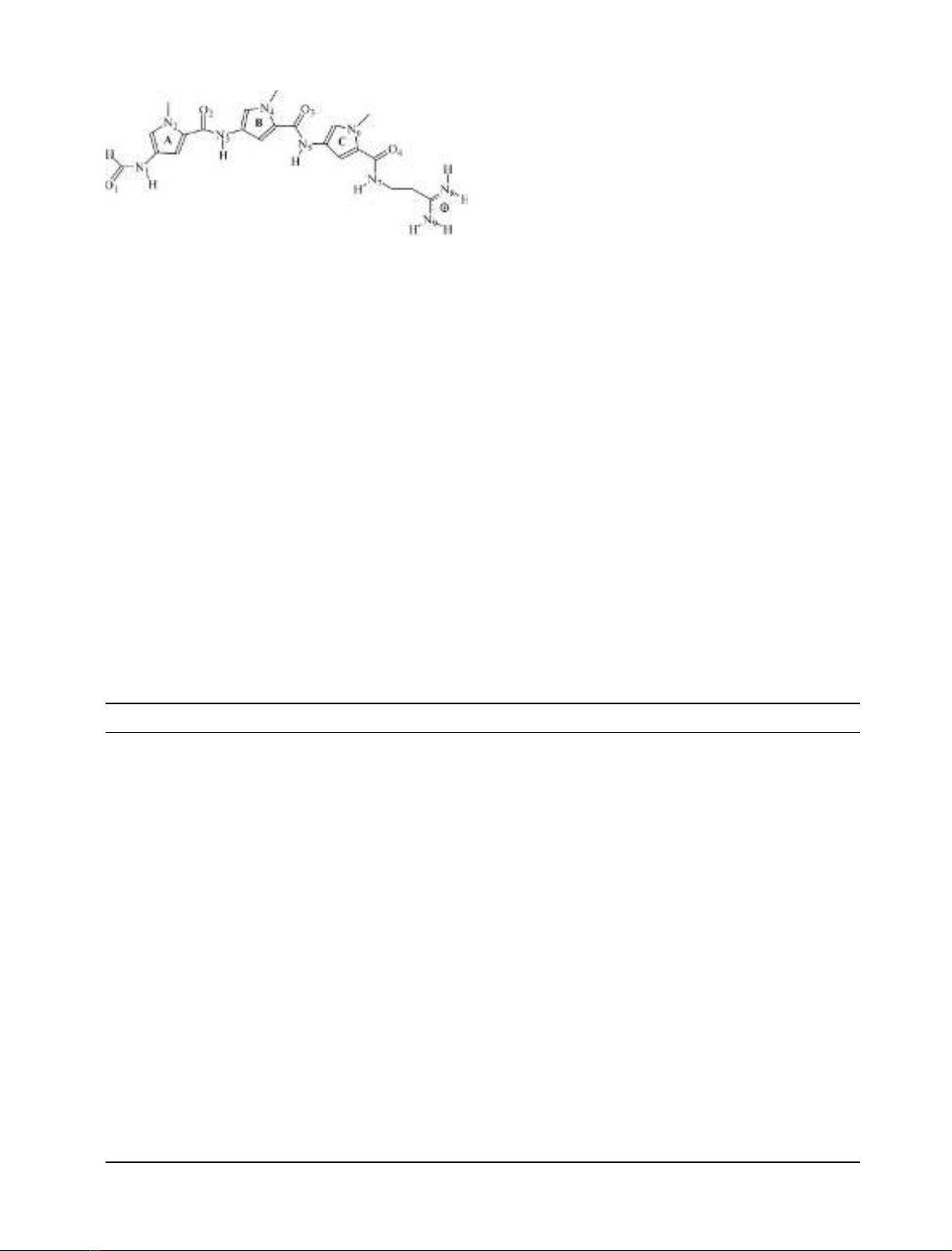
Distamycin A (Fig. 1) is a member of a family of naturally
occuring oligopeptides showing antiviral and antibiotic
properties. Like other minor-groove binder drugs, distamy-
cin binds noncovalently in the minor groove of DNA with a
binding preference for stretches of AT-rich sequences [1],
thereby preventing DNA and RNA synthesis by inhibition
of the corresponding polymerase reaction. The crystal
structure determination of a 1 : 1 distamycin–d(CGCAAA
TTTGCG) complex (12-dista) at 2.2 A
˚resolution shows
that the drug covers five of the six AT base pairs [2]. The
amide nitrogen atoms of the drug form hydrogen bonds to
N3(A) and/or O2(T) atoms in the minor groove. The
complex is further stabilized by van der Waals’ and
electrostatic interactions.
The selectivity for AT-rich sequences of minor-groove
binders was first thought to have sterical reasons: the bulky
NH
2
group at the floor of the minor groove of CG-
containing regions can prevent binding of these drugs [3].
More recently, factors such as minor-groove width influen-
cing the extent of van der Waals’ interactions [4] and
electrostatic interactions between the positively charged
drug and the more negatively charged minor groove in the
case of AT sequences [5] were added.
Solution NMR studies have also discovered side-by-
side binding of two distamycin molecules in the minor
groove of d(CGCAATTGCG) [6]. More structural
information about this 2 : 1 binding mode was first
provided by the crystal structure of d(ICICICIC)–dista-
mycin [7] and later by side-by-side complexes of dista-
mycin with natural targets d(ICITACIC), d(ICATATIC)
and d(GTATATAC) [8,9]. Owing to the overlap of about
75%, the two staggered antiparallel distamycin molecules
span almost eight base pairs and are kept together by
dipole–dipole interactions between stacking pyrrole rings
and amide bonds. Each drug hydrogen-bonds with the
bases of only one DNA strand and stacks with the sugar
rings.
We have previously reported the structure determin-
ation at 1.9-A
˚resolution of the complex of the shor-
ter minor-groove binder 4¢,6-diamidino-2-phenylindole
(DAPI) with d(GGCCAATTGG) (10-DAPI), revealing
a novel off-centered binding with a hydrogen bond
between the drug and a CG base pair [10]. In an attempt
to use similar crystal engineering techniques to improve
the resolution of 1 : 1 distamycin–DNA complexes
(currently 2.2 A
˚for 12-dista and 2.0 A
˚for the dista-
mycin–d(CGCGAATTC
+
GCG) complex where C
+
¼
5-methylcytidine (NDB entry code GDLB41), we have
cocrystallized distamycin with the decamer d(GGCCAA
TTGG). Intensity measurements for two crystals obtained
from different crystallization conditions were carried out
to 2.38 and 1.85 A
˚resolution. Whereas for one crystal the
distamycin orientation and binding site is the same as in
12-dista, the orientation of the drug is inverted in the
other crystal. Both orientations show interactions between
thedrugandguanineNH
2
groups. For the inverted
orientation, the DNA–distamycin interaction is also
characterized by ab initio methods.
Correspondence to L. Van Meervelt, Biomolecular Architecture,
Department of Chemistry, Katholieke Universiteit Leuven,
Celestijnenlaan 200F, B-3001 Leuven (Heverlee), Belgium.
Fax: + 32 16 327990, Tel.: + 32 16 327609,
E-mail: Luc.VanMeervelt@chem.kuleuven.ac.be
Abbreviations: 12-dista, crystal structure of the
d(CGCAAATTTGCG) complex (2); 10-DAPI, crystal structure of
the d(GGCCAATTGG)–DAPI complex (10); MPD, 2-methyl-2,
4-pentanediol; DAPI, 4¢,6-diamidino-2-phenylindole; HF, Hartree-
Fock; MP2, Moeller–Plesset perturbational theory.
Note: a web page is available at
http://www.chem.kuleuven.ac.be/research/bma/
(Received 20 December 2001, revised 10 April 2002,
accepted 23 April 2002)
Eur. J. Biochem. 269, 2868–2877 (2002) FEBS 2002 doi:10.1046/j.1432-1033.2002.02952.x
EXPERIMENTAL PROCEDURES
Crystallization and data collection
The DNA decamer d(GGCCAATTGG) was purchased
from Oswel DNA service (University of Southampton, UK),
distamycin from Serva Biochemica (Heidelberg, Germany).
Crystals were grown at 16 C using the sitting drop method
from two different conditions containing 54.4/33.25 m
M
sodium cacodylate buffer (pH 6.0), 35.0/105.0 m
M
MgCl
2
,
70 m
M
NaCl, 8.8% 2-methyl-2,4-pentanediol (MPD),
10.5 m
M
spermine, 0.25/0.42 m
M
ssDNA and 0.125/
0.21 m
M
distamycin against a 50/35% MPD stock solution.
From a bar-shaped crystal of dimensions 0.4 ·0.1 ·
0.05 mm from condition 1, intensity data were collected at
100 K on a MAR345 imaging plate detector at beamline
X11inanEMBLHamburg(k¼0.9116 A
˚) over a 105 u
range with increments of 1.5 using cryocooling techniques
with a crystal-to-detector distance of 350 mm.
A well-diffracting crystal of dimensions 0.2 ·0.1 ·
0.05 mm from condition 2 was mounted for data collection
at 100 K using a similar protocol at beamline BW7b in an
EMBL Hamburg (150 urange, crystal-to-detector dis-
tance 250 mm, k¼0.8423 A
˚).
Data were processed using the
DENZO
/scalepack [11]
suite of programs. Data collection statistics for both crystals
are given in Table 1. The final resolution limit of the
diffraction pattern was 2.38 A
˚for crystal 1 and 1.85 A
˚for
crystal 2.
Structure solution and refinement
Unit cell parameters and space group indicated isomorph-
ism with the d(GGCCAATTGG)–DAPI structure, which
was used as a starting model (NDB entry code DD0002,
except DAPI and solvent molecules) for further refinement
on F
2
using SHELXL-93 [12]. The nucleotides of strand 1
are labeled G1–G10 in the 5¢fi3¢direction and G11–G20
on strand 2, and the drug is labeled D. After determination
of the weighting factor and positional adjustment of parts of
the DNA structure, water molecules were added, but not in
the minor-groove region. At this stage of the refinement, the
distamycin molecule was located in the (F
o
)F
c
) Fourier
difference map. In subsequent refinement cycles, more water
molecules were gradually added. During the conjugate-
gradient refinement, no torsion angle or hydrogen-bond
restraints were applied. The 1,2 and 1,3 distances used as
dictionary values for distamycin were based on netropsin
[13], except for the formamide end which was based on
fragments retrieved from the Cambridge Structural
Database [14].
Fig. 1. Structure and numbering scheme of distamycin. Hydrogen
atoms attached to pyrrole and alkyl groups are not shown.
Table 1. Data collection and refinement statistics of the d(GGCCAATTGG)–distamycin complexes. NA, Not available.
Crystal 1 Crystal 2
Data collection statistics
Space group P2
1
2
1
2
1
P2
1
2
1
2
1
Unit cell (A
˚)a¼26.011, b ¼40.861, c ¼53.164 a ¼25.289, b ¼36.439, c ¼53.047
Total no. of measured reflections 27524 43223
No. of independent reflections 2538 4223
Resolution (A
˚) 2.38 1.85
Multiplicity 5.5 4.3
v
2
1.035 1.142
R
symm
(%) 4.1 (100.0–2.38 A
˚) 4.9 (100.0–1.85 A
˚)
25.1 (2.42–2.38 A
˚) 19.5 (1.92–1.85 A
˚)
Completeness (%) 92.7 (100.0–2.38 A
˚) 93.0 (100.0–1.85 A
˚)
93.4 (2.42–2.38 A
˚) 96.9 (1.92–1.85 A
˚)
Mean I/r(I) 22.6 21.1
Reflections with I>3 r(I) (%) 80.3 (100.0–2.38 A
˚) 80.3 (100.0–1.85 A
˚)
46.3 (2.42–2.38 A
˚) 55.3 (1.92–1.85 A
˚)
Refinement statistics
Resolution range (A
˚) 100.0–2.38 100.0–1.85
Rvalue/R
free
value (%) 20.30/NA 19.74/27.80
No. of nonsolvent atoms 445 445
No. of water molecules 46 74
Average Bvalues of DNA (A
˚
2
) 66.6 28.2
Average Bvalues of distamycin (A
˚
2
) 79.1 30.8
Average Bvalues of water molecules (A
˚
2
) 73.0 43.0
Rmsd of bond lengths (A
˚) 0.018 0.019
Rmsd of bond angles () 3.20 2.70
FEBS 2002 Two 1 : 1 binding modes for distamycin (Eur. J. Biochem. 269) 2869
For crystal 1, the Rvalue converged to 20.30% after
addition of 46 water molecules (R
free
was not used to avoid
further reduction of the number of data per parameter at
this resolution). For crystal 2, the first maps already
indicated an inverted orientation (hereafter called orienta-
tion B) of the drug molecule with respect to crystal 1
(orientation A). Therefore refinement for crystal 2 was
monitored using R
free
calculated for a reference set of 10%
of the reflections. Addition of 29 water molecules and
distamycin led to the following Rvalues: R¼23.88%,
R
free
¼32.31% for orientation A, and R¼22.79%,
R
free
¼30.75% for orientation B. Final Rvalues for crystal
2wereR¼19.74%, R
free
¼28.01% for orientation B. An
independent refinement with orientation A resulted in
R¼20.30%, R
free
¼29.84%. As a consequence and in
agreement with the electron density maps, orientation B was
retained for crystal 2.
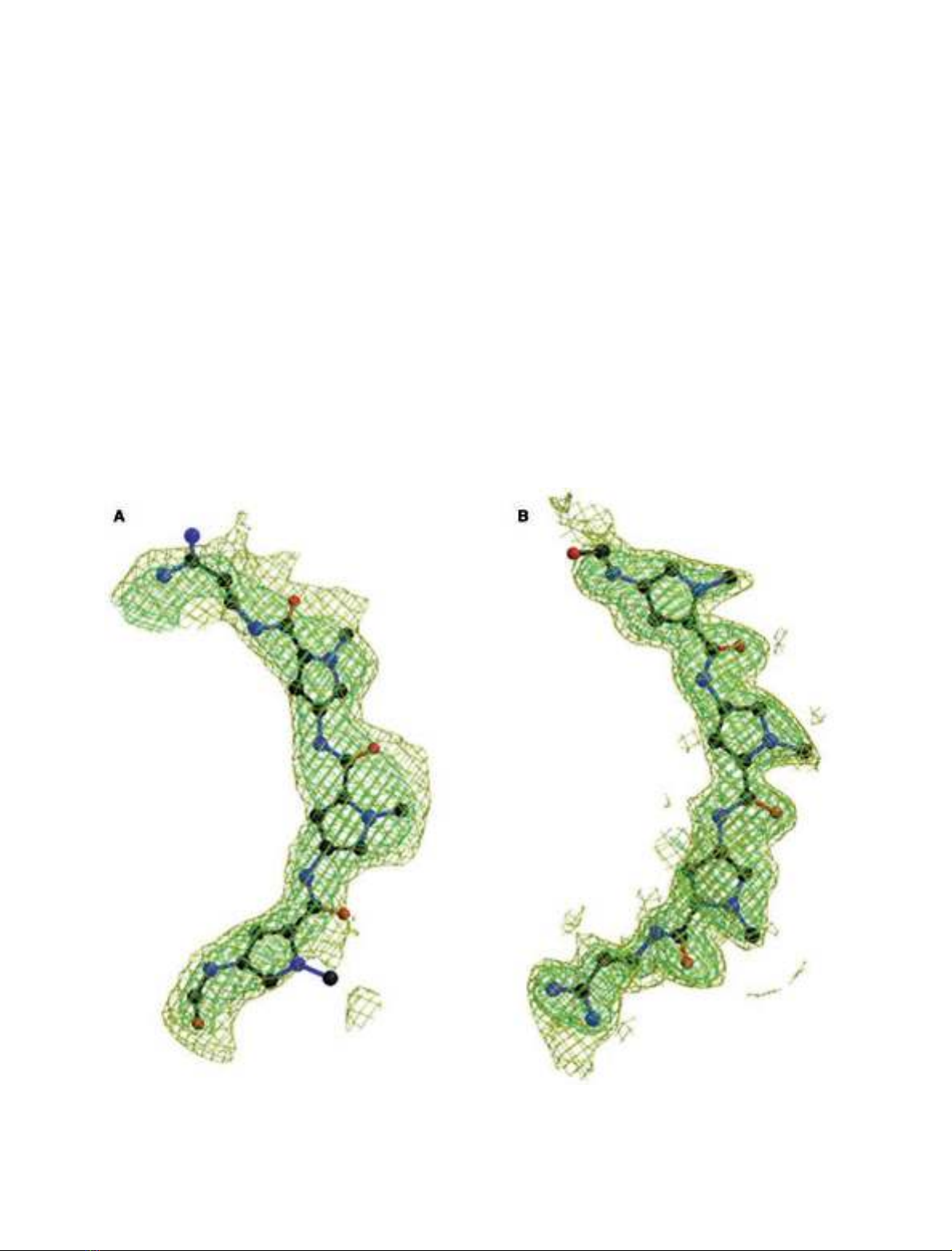
Figure 2 shows the final (2F
o
)F
c
) electron-density maps
in the minor-groove region for both crystals. Refinement
statistics are presented in Table 1.
The helical parameters in accordance with the Tsukuba
Workshop guidelines [15], and torsion angles were calcula-
tedwiththe3
DNA
program [16].
Quantum chemical calculations
Ab initio quantum chemical calculations were used to
investigate intrinsic molecular interactions of a number of
close intergroup contacts observed in the crystal. Special
attention was given to contacts involving the guanine amino
groups. Appropriate fragments of the drug and several
proximal bases have been taken from the crystal structure.
Intermolecular positions of the interacting species were
frozen based on the crystal data using a set of constraints
involving three (in some cases more) appropriate non-
hydrogen atoms on each monomer. The rest of the structure
including all the hydrogen positions was relaxed using
gradient optimization. This procedure has been extensively
used in the past to investigate local contacts seen in DNA
crystal structures and allows full relaxation of the electronic
structure and hydrogen positions while keeping the systems
Fig. 2. Final (F
o
)F
c
) electron-density maps in the minor groove of the crystal structure of the d(GGCCAATTGG)–distamycin complex in which the
drug has been omitted from the final refined model. (A) Crystal 1 in orientation A; (B) crystal 2 in orientation B. The refined distamycin position is
superimposed on the difference density for reference, contouring at 1 r(yellow) and 2 rlevel (green). The figure was prepared with
BOBSCRIPT
[34]
and
RASTER
3
D
[35].
2870 K. Uytterhoeven et al.(Eur. J. Biochem. 269)FEBS 2002

studied in the experimental geometry [10,17–20]. The
optimizations were carried out within the Hartree–Fock
(HF) approximation with the standard polarized 6–31G*
basis set. Although this level of calculations underestimates
the flexibility of amino groups, it nevertheless is sufficient to
reveal when the amino group is activated by molecular
interactions towards a partial sp
3
hybridization [21–23].
Interaction energy calculations between drug fragments
and nucleobases were carried out assuming the quantum
chemical-optimized geometries with inclusion of electron
correlation effects using the second-order Moeller-Plesset
perturbational theory (MP2) with the 6–31G basis set
augmented by diffuse d-polarization functions to all sec-
ond-row elements [exponents of 0.25, designated as
6–31G*(0.25)] to properly account for the dispersion
attraction [22,23]. The calculations were corrected for the
basis set superimposition error using the standard counter-
poise procedure. The quantum chemical procedure used in
this study allows reliable semiquantitative characterization
of the nature and intrinsic in vacuostrength of the observed
intermolecular contacts. All quantum chemical calculations
were carried out using the
GAUSSIAN
94 program suite [24].
RESULTS AND DISCUSSION
Both complexes (Fig. 3) have a conformation that closely
resembles the native decamer structure [25] and its DAPI
complex [10]: a central octamer d(CCAATTGG) consisting
of normal Watson–Crick base pairs is at both ends flanked
by two overhanging guanines forming triplets in the crystal
packing. All three drug–decamer complexes differ from the
native 1.15-A
˚resolution structure in the phosphate confor-
Fig. 3. Stereoscopic representation of the
distamycin–d(GGCCAATTGG) complexes.
(A) Crystal 1 in orientation A; (B) crystal 2 in
orientation B. The DNA is drawn with open
bonds, and the distamycin with solid bonds.
Nitrogen, oxygen and phosphor atoms are
grey, and carbon atoms are white. The figure
was prepared using B
OBSCRIPT
[34].
FEBS 2002 Two 1 : 1 binding modes for distamycin (Eur. J. Biochem. 269) 2871
mation of residue C13, which correlates with the difference
in resolution as described previously [10]. As expected for
B-DNA, the sugar puckering modes for the central octamer
duplex are situated in the normal C3¢-exo to O4¢-endo
range, the southern part (C2¢-endo) of the pseudorotation
cycle. However, for the overhanging guanines, C3 and C13,
the puckering modes are closer to those for A-DNA, which
is a consequence of triplet formation.
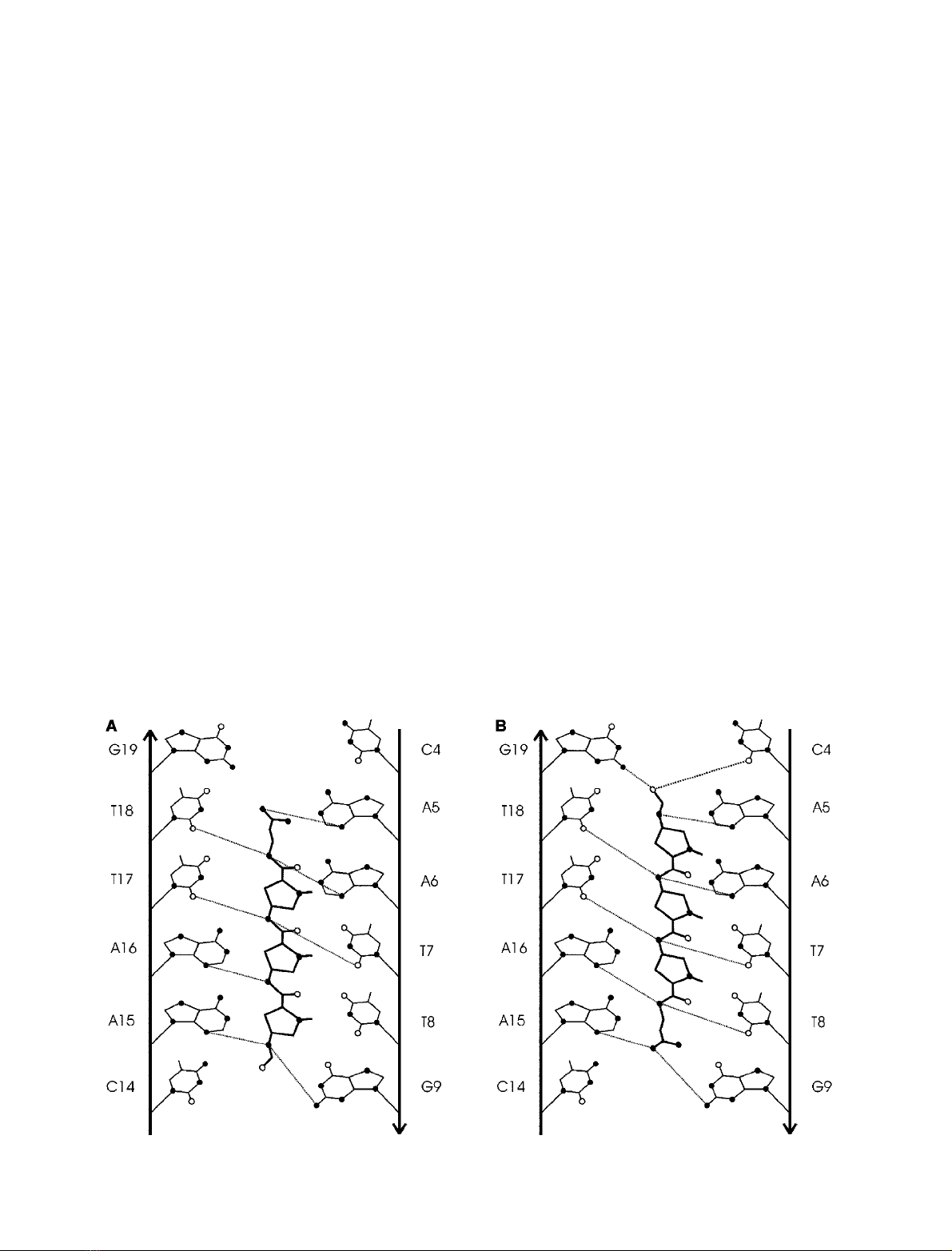
DNA–distamycin interactions
Distamycin binds in the minor groove of the central
octamer. The expected binding site of at least five base
pairs makes interactions with GC base pairs inevitable.
Both structures not only differ in resolution, but also in the
orientation of the distamycin molecule (Fig. 4). In the lower
resolution structure of crystal 1, the orientation (orienta-
tion A) is similar to that described for 12-dista [2]. The
higher-resolution structure of crystal 2 shows an inverted
orientation of distamycin (orientation B).
Position of distamycin in the lower-resolution structure
(orientation A, Fig. 4A)
Distamycin binds to five base pairs covering the sequence
d(AATTG). The positively charged amidinium end group is
orientated toward the A5.T18 base pair with atom N9(D)
lying deep in the minor groove interacting with N3(A5)
(2.70 A
˚) and the partial negative O4¢atoms O4¢(A5)
(3.29 A
˚)andO4¢(A6) (2.80 A
˚). A close contact with the
amino group of G19 is avoided (N9(D)…N2(G19) 3.86 A
˚).
The nearest neighbor of N8(D) is O4¢(3.41 A
˚). Bifurcated
hydrogen bonds are formed between the amide nitrogen
atoms simultaneously to both N3(A6) and O2(T18) for
N7(D), O2(T7) and O2(T17) for N5(D), and N3(A15) and
N2(G9) for N1(D). N3(D) only has one close contact to
N3(A16) and is 3.59 A
˚away from O2(T8). At the other end
of distamycin, the formamide O1(D) is oriented away from
the groove. The nearest neighbor of O1(D) is a sugar
O4¢(A15) atom at 3.50 A
˚.
The three pyrrole rings are rotated to each other to
follow closely the curvature of the groove. Rings A and B
make an angle of 22.9 , rings B and C 28.4 and ring C
makes an angle of 35.9 with the terminal amidinium
group. The drug–DNA complex is further stabilized by
van der Waals’ interactions mainly between carbon atoms
of the three pyrrole rings and the sugar–phosphate
backbone. No water molecules are in direct contact with
the drug molecule.
Position of distamycin in the higher-resolution
structure (orientation B, Fig. 4B)
It was clear from the density maps of the 1.85-A
˚resolution
structure that the drug has to be rotated over 180 with
respect to orientation A. As a consequence, the amidinium
group is now orientated toward the G9–C12 base pair and
distamycin now interacts with six base pairs.
The N9(D) atom of the amidinium group is in close
contact with amino group N2 of guanine G9 (3.16 A
˚)and
hydrogen-bonded with N3(A15) (2.85 A
˚). This N9(D) atom
is also in contact with O2(C14) by two intermediate water
molecules.
The O1(D) atom of the formamide group now points
toward the C4–G19 base pair with a very close contact with
N2(G19)ofonly2.57A
˚. Atom O1(D) is further in contact
with O2(C4) (3.39 A
˚), O4¢(A5) (2.94 A
˚)andtwowater
molecules (2.77 and 2.95 A
˚). In the same formamide group,
atom N1(D) interacts with N3(A5) (3.12 A
˚)andO4¢(A6)
(3.03 A
˚).
Fig. 4. Schematic view of the distamycin interactions in the minor groove of d(GGCCAATTGG)–distamycin structures. (A) Crystal 1; (B) crystal 2.
2872 K. Uytterhoeven et al.(Eur. J. Biochem. 269)FEBS 2002


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)