
Functional epitope of common cchain for interleukin-4 binding
Jin-Li Zhang, Manfred Buehner and Walter Sebald
Theodor-Boveri-Institut fu
¨r Biowissenschaften (Biozentrum), Physiologische Chemie II, Universita
¨tWu
¨rzburg, Germany
Interleukin 4 (IL-4) can act on target cells through an IL-4
receptor complex consisting of the IL-4 receptor achain and
the common cchain (c
c
). An IL-4 epitope for c
c
binding has
previously been identified. In this study, the c
c
residues
involved in IL-4 binding were defined by alanine-scanning
mutational analysis. The epitope comprises c
c
residues I100,
L102, and Y103 on loop EF1 together with L208 on loop
FG2 as the major binding determinants. These predomin-
antly hydrophobic determinants interact with the hydro-
phobic IL-4 epitope composed of residues I11, N15, and
Y124. Double-mutant cycle analysis revealed co-operative
interaction between c
c
and IL-4 side chains. Several c
c
residues involved in IL-4 binding have been previously
shown to be mutated in X-linked severe combined
immunodeficiency. The importance of these binding residues
for c
c
function is discussed. These results provide a basis for
elucidating the molecular recognition mechanism in the IL-4
receptor system and a paradigm for other c
c
-dependent
cytokine receptor systems.
Keywords:commoncchain; interleukin 4; mutagenesis;
protein–protein interaction; structure/function.
Interleukin-4 (IL-4) is a multifunctional cytokine that plays
a critical role in the regulation of immune responses [1,2]. It
induces the generation of Th2-dominated early immune
response [3] and determines the immunoglobulin class
switching to IgE [4]. Dysregulation of IL-4 function is
strongly correlated with type I hypersensitivity reactions,
such as allergies and asthma [5]. The IL-4 receptor complex
is therefore a potential target for the development of
antiallergic drugs. The central role of IL-4 in the develop-
ment of Th2 cells suggests that it may be of benefit in the
treatment of autoimmune disease characterized by an
imbalance of Th cells [6]. Its ability to induce growth arrest
and apoptosis in leukemic lymphoblasts in vitro [7] suggests
that IL-4 is also a promising cytokine for the treatment of
high-risk acute lymphoblastic leukemia. Understanding the
molecular recognition mechanism in the IL-4 receptor
system is a prerequisite for the rational design of IL-4-like
drugs.
IL-4 is one of the short-chain four-helix bundle cytokines.
Its effects depend on binding to and signaling through a
receptor complex consisting of a primary high-affinity
binding subunit, the IL-4Ra, and a low-affinity receptor,
depending on the cell type, the common cchain (c
c
;typeI
IL-4 receptor [8]) or IL-13Ra1 chain (type II IL-4 receptor
[9]). All three receptors are members of the type I cytokine
receptor superfamily, which is characterized by the presence
of at least one cytokine-binding homology region (CHR)
composed of two fibronectin type III domains. The
membrane distal domain contains a set of four conserved
cysteines, and the membrane proximal domain contains a
WSXWS motif [10]. The fibronectin type III domain is
comprised of seven bstrands, the sequences of which are
conserved between members of the family, while loop
sequences connecting the bstrands vary between family
members and putatively contain residues that mediate
distinct intermolecular contacts. These loop regions were
therefore selected for this mutational analysis.
A comprehensive mutational analysis of IL-4 in which
single residues were replaced by alanine or charged residues
yielded high-resolution data on the binding epitopes for the
receptor chains. The IL-4 site 1 binding epitope for IL-4Ra
consists of a mixed charge pair (E9, R88) as major
determinants and five minor determinants located on helices
A, B, and C [11]. The importance of site 1-binding
determinants and their partner residues on IL-4Ra(D72,
Y183 as key binding determinants) was subsequently
confirmed and further defined by determining the crystal
structure of the 1 : 1 IL-4/IL-4Raectodomain (IL-4-
binding protein, IL-4BP) complex [12] and by mutational
analysis of the IL-4BP binding epitope [13]. The results have
already been used for the rational design of IL-4 minipro-
teins [14]. The IL-4 site 2 epitope for c
c
comprises residues
I11 and N15 on helix A together with Y124 on helix D as
major binding determinants and three minor determinants
K12, R121, and S125 on helices A and D [15]. A double
mutant of IL-4 that completely inhibits responses induced
by IL-4 and IL-13 by disrupting the binding of the IL-4 site
2 epitope to c
c
or IL-13Ra1provedtobeaverypromising
anti-asthma drug [16–18]. Two further IL-4 mutants that
selectively inhibit IL-4-induced activity on endothelial cells
appeared to be good candidate drugs for the treatment of
certain autoimmune diseases [6] and high-risk acute
lymphoblastic leukemia [7]. However, the residues on c
c
that contribute to IL-4 site 2 binding remain uncertain.
Correspondence to W. Sebald, Theodor-Boveri-Institut fu
¨rBiowis-
senschaften (Biozentrum), Physiologische Chemie II, Universita
¨t
Wu
¨rzburg, Am Hubland, D-97074 Wu
¨rzburg, Germany.
Fax: + 49 931 888 4113, Tel.: + 49 931 888 4111,
E-mail: sebald@biozentrum.uni-wuerzburg.de
Abbreviations: IL-4, interleukin-4; IL-4Ra, interleukin-4 receptor a
chain; IL-4BP, IL-4 binding protein; c
c
, common cchain; IL-13Ra1,
IL-13 receptor a1 chain; CHR, cytokine-binding homology region;
Jak, Janus kinase; XSCID, X-linked severe combined immunodefi-
ciency; hGHR, human growth hormone receptor; hEPOR, human
erythropoietin receptor; b
c
,commonbchain.
(Received 14 November 2001, revised 16 January 2002, accepted
21 January 2002)
Eur. J. Biochem. 269, 1490–1499 (2002) ÓFEBS 2002

c
c
is shared by several important cytokine receptor
complexes, including those for IL-2, IL-4, IL-7, IL-9, IL-15
[8] and also for the recently described new member of the
cytokine family, IL-21 [19]. c
c
alone binds ligands with very
low affinity (K
d
150 l
M
for IL-4) [15]. Recruitment of c
c
into receptor complexes for the above cytokines increases
receptor affinity for binding [20–22]. c
c
participates in
cytokine signaling in several receptor complexes via JAK3
[23]. Mutations of either c
c
or JAK3 result in X-linked
severe combined immunodeficiency (XSCID) which is
characterized by a failure in T and NK cell development
[24]. c
c
-knockout mice have been generated and their
immune system successfully reconstituted by gene therapy
[25,26]. Initial attempts at gene therapy for patients with
XSCID had been successful for more than 10 months
[27,28]. Thus, defining the IL-4-binding determinants on c
c
is important not only for elucidating molecular recognition
and activation mechanisms in the IL-4 receptor system and
possibly providing a paradigm for other c
c
-dependent
cytokine receptor systems, but also for delineating the
molecular pathology of XSCID.
So far, the binding epitopes of human and murine c
c
for some c
c
-dependent cytokines have been studied.
A molecular mapping study using the antagonistic
monoclonal antibody PC.B8, which reacts with a discon-
tinuous site on human c
c
, localized c
c
binding residues to
four loops, but did not identify single specific residues for
ligand binding [29]. Mutational analysis of murine c
c
employing heterodimeric IL-2R and IL-7R on whole cells
suggests that c
c
epitopes for IL-2 and IL-7 binding
overlap and comprise at least three distinct putative loop
segments of the c
c
protein [30]. Here we report the effect
of single amino-acid substitutions in the human c
c
ectodomain on IL-4 binding. Biosensor techniques
employing soluble recombinant IL-4, IL-4-BP and the
wild type or mutant forms of human c
c
ectodomain
revealed the contributions of c
c
residues to IL-4 binding.
The possible co-operativity between some residues on the
c
c
epitope and the IL-4 site 2 epitope was analyzed by
double-mutant cycle analysis.
EXPERIMENTAL PROCEDURES
Protein expression and purification
The ectodomain of human c
c
comprising amino-acid
residues 1–232 [20] was expressed with a C-terminal
thrombin cleavage site (LVPRGS) plus a His
6
tag in SF9
insect cells according to the manufacturer’s instructions
(PharMingen). The protein was isolated from the culture
medium of infected SF9 cells by standard procedures
involving Ni
2+
/nitriloacetate/agarose (Qiagen), digested
with thrombin (Sigma), and purified by gel-filtration
chromatography through a Superdex 200 HR 10/30 col-
umn (Pharmacia). After exhaustive dialysis against water,
the purified protein was freeze-dried and stored at )80 °C.
ThecDNAforthemurinec
c
ectodomain comprising
residues 1–233 [31] was cloned into the temperature-
regulated expression vector pRpr9 fd [32], expressed in
Escherichia coli strain KS 474, and refolded as described
[33]. The refolded protein was purified to homogeneity by
gel-filtration chromatography through a Superdex 200 HR
10/30 column, and stored at )80 °C.
The A182, C207 IL-4BP variant was produced in SF9
cells, purified, and biotinylated at C207 as described [32].
IL-4 and IL-4 variants were expressed in E. coli, refolded,
and purified to homogeneity as described [11,34]. Protein
concentrations were determined by measuring A
280
,using
an absorption coefficient (e
280
)¼8860
M
)1
Æcm
)1
for
IL-4, e
280
¼7370
M
)1
Æcm
)1
for A124 IL-4, e
280
=
66 930
M
)1
Æcm
)1
for IL-4BP, e
280
¼61 450
M
)1
Æcm
)1
for
human c
c
,e
280
¼60 170
M
)1
Æcm
)1
for A103 human c
c
,
and e
280
¼45 660
M
)1
Æcm
)1
for murine c
c
.
Mutagenesis of the c
c
ectodomain
cDNA for human c
c
ectodomain was submitted to in vitro
cassette mutagenesis employing synthetic double-stranded
oligonucleotides. The c
c
variants were expressed and
purified as the wild-type human c
c
ectodomain.
Biosensor interaction analysis
The binding of c
c
variants to IL-4/IL-4BP was recorded on a
BIAcore 2000 system (Pharmacia Biosensor) as described
[15]. Briefly, a CM5 biosensor chip was first loaded with
streptavidin in flow cells 1 and 2. Subsequently biotinylated
A182,C207 IL-4BP was immobilized at the streptavidin
matrix of flow cell 2 at a density of 200 resonance units.
The following reaction cycle was applied using the com-
mand COINJECT: (a) IL-4 at 0.1 l
M
in HBS buffer (10 m
M
Hepes, pH 7.4, 150 m
M
NaCl, 3.4 m
M
EDTA, 0.005%
surfactant P20) was perfused over flow cells 1 and 2 at a flow
rate of 10 lLÆmin
)1
at 25 °C for 2 min; (b) 0.1 l
M
IL-4 plus
c
c
ectodomain or c
c
variants at 1–10 l
M
inthesamebuffer
were perfused in the same way for 2 min; (c) HBS buffer
alone was perfused for 5 min; (d) free receptors were
regenerated by perfusion with 0.1
M
acetic acid/1
M
NaCl
for 30 s. Sensograms were recorded at a data-sampling rate
of 2.5 Hz and evaluated as described [15]. Equilibrium
binding of c
c
variants at 1, 2, 3, 5, 10 l
M
was measured for at
least three times in duplicate. The mean standard deviation
(mean r) was 13.8% ± 6.5% for the K
d
values calculated
from the five variant concentrations. For the double mutant
cycle analysis [35], the same procedure as above was used
except that IL-4 variants [15,36] at 0.1 l
M
and c
c
variants at
2, 4, 6, 10, 20 l
M
were perfused (the mean rwas
16.4% ± 7.4% for the K
d
values). The loss of binding free
energy on mutation for IL-4 and c
c
wascalculatedasddG
(kJÆmol
)1
)¼5.69 log K
d
(mutant)/K
d
(wild-type). The
interaction energy between two residues was calculated by
the double-mutant cycle method as in Eqn. (1):
ddGint ¼ddGX-AþddGY-BÿddGX-A;Y-Bð1Þ
where ddG
X-A
and ddG
Y-B
are the changes in binding
energy on mutation of X to A and Y to B (mutation of IL-4
and c
c
in this experiment), respectively, and ddG
X-A, Y-B
the
change on the simultaneous mutation of X to A and Y to B.
ddG
int
is a measure of the co-operativity of the interaction
of the two components mutated. ddG
int
¼0indicatesthat
the pair of residues analyzed do not interact. A positive
value of ddG
int
means that two residues interact favorably,
and a negative value means that the two residues repel each
other [37]. The individual errors (2 r,a¼0.95) calculated
from the mean for ddG
int
areshowninTable3.
ÓFEBS 2002 Mutagenesis of human c
c
ectodomain (Eur. J. Biochem. 269) 1491
Molecular modeling of the IL-4–IL-4BP–c
c
ternary
complex
The present model is based on the crystal structure of the
complexofIL-4andIL-4BP(PDBentry1IAR[12]),
augmented by the model of c
c
derived from human growth
hormone receptor (hGHR), as obtained from an older
model (T. Mueller, & W. Kammer, personal communica-
tion, Universita
¨tWu
¨rzburg, Germany)
1of the ternary
complex of IL-4–IL-4BP–c
c
. This old model was based on
the structure of free IL-4 (PDB entry 1HIK [38]) and of
models of the extracellular domains of IL-4Raand c
c
obtained by analogy modeling following the structure of the
hGHR complex (PDB entry 3HHR [39]). The 3HHR data
were obtained from the protein databank (PDB [40]). The
old model was built in such a way that all cysteine residues
formed proper disulfide bonds, and all evidence from
mutation experiments available at the time was used to
adjust the binding epitopes of the receptor chains. The
resulting alignment required some nontrivial rebuilding with
insertions and deletions, and, consequently, the resulting
model of the IL-4 receptor complex had to be extensively
energy refined. The program
O
[41] was used for model
building, and the program
X
-
PLOR
[42] for energy refinement.
The differences between the experimentally determined
binary complex and the corresponding components of the
old model were significant in detail, but the gross changes
were small enough that the binding topology of c
c
could be
transferred to the new model without major problems.
The local program
DISDM
2 was used (H. J. Hecht, &
M. Buehner, unpublished results) to build and adjust the
present model using the data of mutational analysis of IL-4
and c
c
. The program runs under Open-VMS and uses
Datagraph VTC 8002 and VTC 8003 terminals for display.
All model building was performed manually. For online
refinement of conformational energy, the program
EREF
was used [43], which is called from within
DISDM
2.
RESULTS
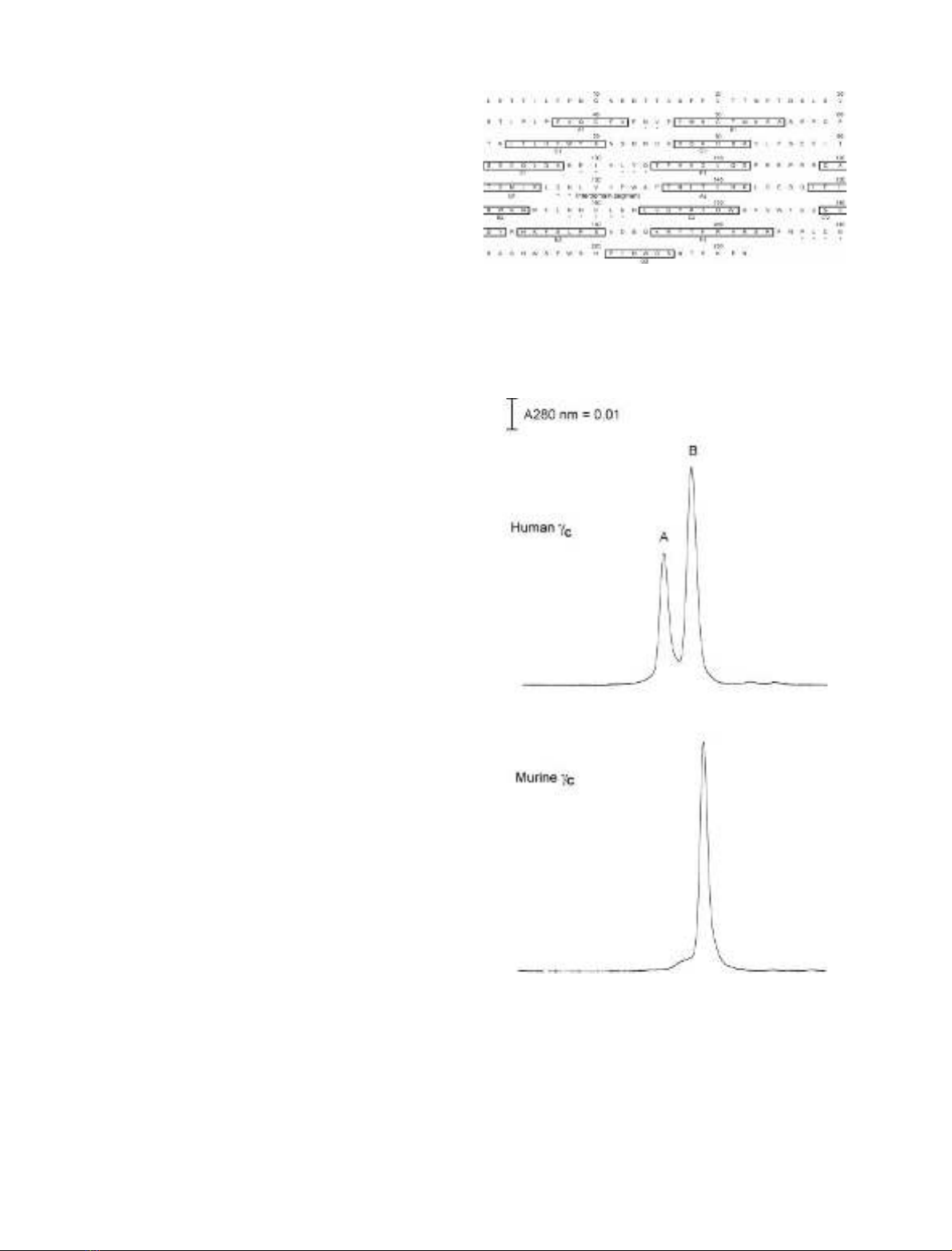
Site-specific mutagenesis of amino acids
in the c
c
ectodomain
Alanine substitutions were targeted to residues in four
putative interconnecting loops and the interdomain segment
of the human c
c
ectodomain based on the published models
[44–46], and sequence alignment performed between c
c
and
several cytokine receptors, the major ligand-binding deter-
minants of which were identified. These include the hGHR
[39,47], the human erythropoietin receptor (hEPOR
[48,49]), IL-4BP [12], and the human gp130 (hgp130
[50,51]). Eighteen c
c
variants were generated with amino-
acid substitutions in the AB1, EF1, BC2, FG2 loops and the
interdomain segment (Fig. 1). A deletion mutant lacking
residues 1–33 of the N-terminus of c
c
,namedc
c
CHR, was
also generated to find out whether this N-terminal region of
c
c
is required for ligand binding.
All human c
c
wild-type or variant proteins could be
purified to apparent homogeneity by Ni
2+
/nitrilotriacetate/
agarose and gel filtration. The wild-type human c
c
ectodo-
main expressed in SF9 cells was recovered as monomeric
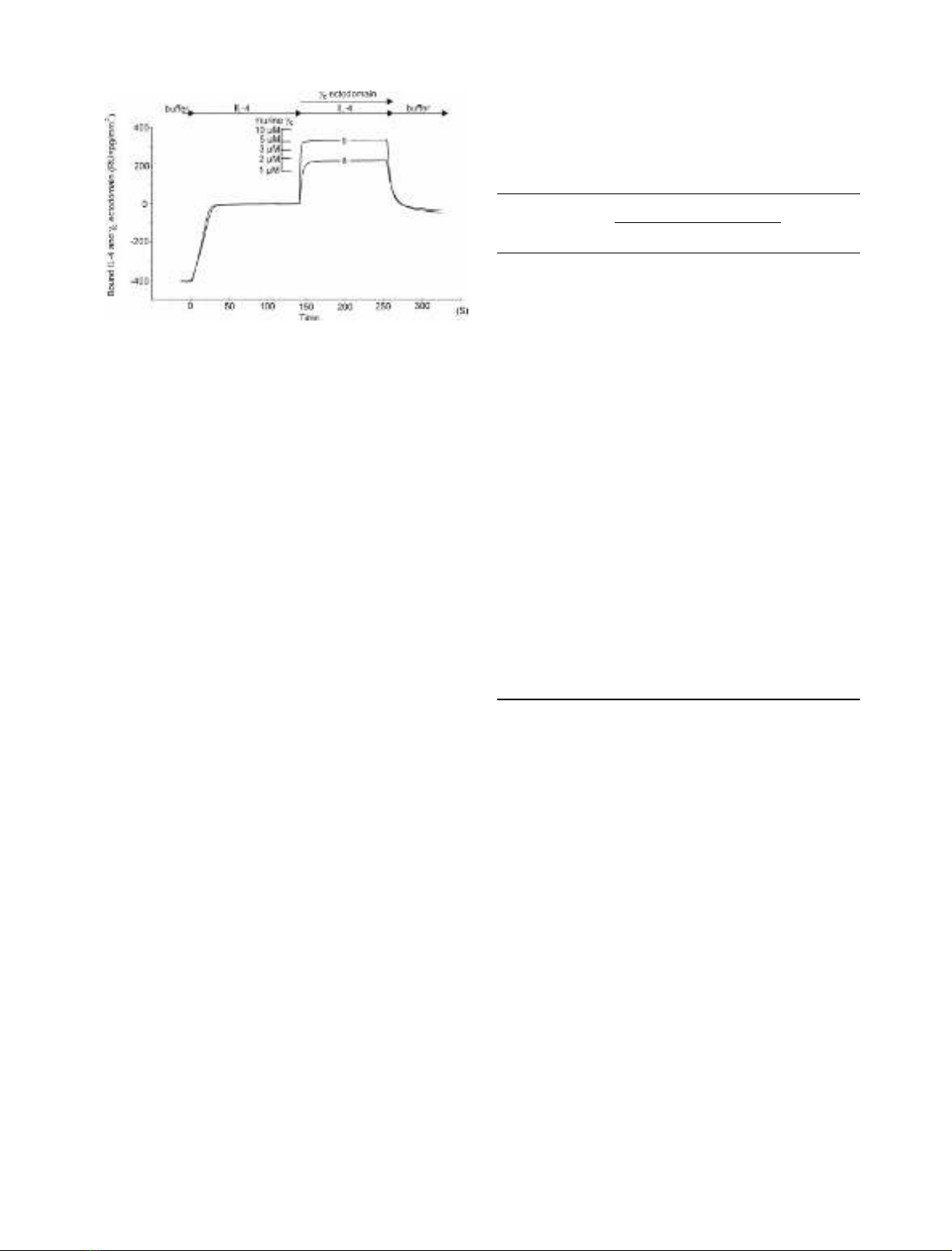
and dimeric species [52,53]. The murine c
c
ectodomain
expressed in E. coli occurred as a monomer (Fig. 2). Initial
biosensor studies showed that the different forms of human
and murine proteins exhibited similar binding affinity for
the IL-4–IL-4BP complex. The mixture of monomeric and
dimeric human c
c
interacts with the complex with a K
d
of
Fig. 1. Amino-acid substitutions in the ectodomain of the human com-
mon cchain (c
c
). The amino-acid sequence of c
c
is shown with boxed
portions indicating predicted b-strands which are designated by the
letter below the box. Residues substituted in this study are indicated by
asterisks.
Fig. 2. Gel-filtration analysis of the human c
c
ectodomain expressed in
SF9 cells and the murine c
c
ectodomain expressed in E. coli. The samples
were applied to a Superdex 200 HR 10/30 column and eluted with the
same buffer. The two peaks of human c
c
represent dimer (A) and
monomer (B).
1492 J.-L. Zhang et al.(Eur. J. Biochem. 269)ÓFEBS 2002
4l
M
, and murine c
c
with a K
d
of 1.6 l
M
(Fig. 3 and
Table 1). In addition, different preparations of wild-type
human c
c
ectodomain consistently showed a K
d
of 4 l
M
irrespective of the monomer to dimer ratio (data not
shown). Therefore, the mixtures of dimeric and monomeric
human c
c
protein were used for all biosensor measurements.
The c
c
epitope for IL-4 binding
The method of measuring the binding of c
c
to IL-4–IL-4BP
by biosensor was established previously [15]. The dissocia-
tion constant K
d
evaluated from the concentration
dependence of equilibrium binding proved to be very
reliable for measuring the interaction of the c
c
ectodomain
with IL-4–IL-4BP. The measured K
d
for interaction of c
c
ectodomain variants with IL-4–IL-4BP are compiled in
Table 1. Eight c
c
variants including c
c
CHR exhibited
unchanged binding characteristics. Changes in binding
affinity were observed in 11 c
c
variants. The K
d
of six
variants was too high to be reliably determined. A rough
estimate yields K
d
values of about 200–300 l
M
for I100A,
L102A, Y103A and L208A, and K
d
values of about 500–
1000 l
M
for C160A and C209A. The K
d
values of five
variants, N128A, H159A, L161A, E162A, and G210A,
were found to be increased threefold to fourfold compared
with the K
d
of wild-type c
c
, suggesting that these residues are
part of the c
c
binding interface, but do not play a key role in
binding. The loss of binding affinity of the four variants
I100A, L102A, Y103A and L208A is not likely to be caused
by extended structural alterations, as I100A, L102A, and
L208AwerereportedtobindtoIL-2andIL-7withthe
same affinity as wild-type c
c
, and the Y103A mutation
resulted in only twofold to threefold reduced IL-2 and IL-7
binding [30]. Thus, the four residues I100, L102, Y103 and
L208 are hot spots on c
c
, contributing > 9 kJÆmol
)1
each.
The five minor residues investigated contribute only 2.9–
3.5 kJÆmol
)1
. The two cysteine variants C160A and C209A
exhibited a largely reduced binding affinity (K
d
> 500 l
M
).
Thismaybecausedbystructuralperturbationofthe
protein. A direct role in binding, however, cannot be
excluded for these residues.
Double-mutant cycle analysis of the IL-4–c
c
interface
The co-operativity of the interaction of some residues on the
IL-4 site 2 epitope and the c
c
ectodomain was determined in
this experiment. Double-mutant cycles were constructed
only for the mutants with minimal effects on binding
(Tables 2 and 3), because the K
d
values for the interaction
between variants of the main binding residues I100, L102,
Y103, and L208 of c
c
andIL-4variantsaswellasthe
interaction between variant I11A of IL-4 and c
c
variants
were too high to be reliably determined. Interaction between
residues on IL-4 and c
c
can be grouped according to their
coupling energies (Table 3). The main binding determinant
of IL-4 Y124 failed to exhibit positive coupling energies with
any of the c
c
residues analyzed. IL-4 Y124 probably
interacts with the main functional side chains of the receptor
located on loop EF1 (I100, L102, Y103), the binding of the
alanine variants of which was too weak to be analyzed by
this approach. Remarkably, IL-4 S125 neighboring Y124
does show coupling to receptor N128 in addition to that to
Fig. 3. Sensograms recording the binding of human and murine c
c
ectodomains to the IL-4–IL-4BP complex. IL-4BP was immobilized on
the biosensor matrix. At time zero, perfusion with 100 n
M
IL-4 was
initiated. The saturation binding of IL-4 was arbitrarily set as zero.
After 120 s, perfusion was continued with 100 n
M
IL-4 plus c
c
ecto-
domain. In different cycles, 5 l
M
human (a) or murine (b) c
c
ecto-
domains were applied. Perfusion with buffer alone started at time
240 s. The ruler indicates resonance units (RU) corresponding to
1–10 l
M
murine c
c
ectodomain. The resonance unit for 5 l
M
human c
c
corresponded to that for 1.6–2 l
M
murine c
c
.
Table 1. Equilibrium binding between c
c
ectodomain mutants and
IL-4–IL-4BP. The dissociation constants K
d
were evaluated from
equilibrium binding between wild-type (wt) or mutants (mut) of the c
c
ectodomain and immobilized IL-4BP saturated with IL-4. The loss of
free energy of binding on mutation was calculated as ddG
(kJÆmol
)1
)¼5.69 log K
d
(mut)/K
d
(wt).
Alanine
variant
Equilibrium binding
ddG
(kJÆmol
)1
)
K
d
(l
M
)K
d
(mut)/K
d
(wt)
Murine c
c
(wt) 1.6
Human c
c
(wt) 4.0 1.0 0.0
Human c
c
CHR 4.5 1.1 0.2
Loop 1 (AB1)
N44A 2.8 0.7 )0.9
V45A 3.3 0.8 )0.5
Loop 3 (EF1)
E99A 5.5 1.4 0.8
I100A >320 >80 >11
L102A >240 >60 >10
Y103A >300 >80 >11
Q104A 5.6 1.4 0.8
Loop 4 (ID)
Q127A 2.4 0.6 )1.3
N128A 15 3.7 3.2
Loop 5 (BC2)
N158A 1.4 0.4 )2.3
H159A 17 4.1 3.5
C160A >900 >230 >13
L161A 13 3.2 2.9
E162A 13 3.2 2.9
Loop 6 (FG2)
P207A 3.4 0.9 )0.4
L208A >166 >40 >9
C209A >490 >120 >12
G210A 15 3.7 3.2
ÓFEBS 2002 Mutagenesis of human c
c
ectodomain (Eur. J. Biochem. 269) 1493

G210. The IL-4 side chain of N15 functionally interacts with
the central receptor side chain N128, and also with H159
located at the periphery of the functional c
c
epitope. The
relative positions of the coupling side chains as proposed by
our theoretical model of the ternary complex (see below) are
presented in the open-book view in Fig. 4A,B. The two
receptor side chains N1128 and H159 are 12 A
˚apart in the
c
c
model. This could indicate that our c
c
model is
inaccurate, because this model does not completely fit the
results of the double-mutant cycle analysis. Alternatively,
the interaction of IL-4 side chain N15 with H159 (coupling
energy only 0.8 kJÆmol
)1
) may be indirect. Of particular
interest is the IL-4 side chain of R121, which, after being
substituted with D or E, leads to a selective IL-4 agonist
specifically impaired in IL-13Ra1 binding [6,7,16,17,34].
The IL-4 R121 was distinct in showing positive coupling
during interaction with the c
c
side chain L161.
Model of the structure of the IL-4–IL-4BP–c
c
ternary
complex
In a series of steps, c
c
was adapted to achieve a good fit to
the core structure (the binary complex; Fig. 5). The
procedure started with moving the whole chain (Ôrigid
bodyÕ). Then domains and subdomains were moved indi-
vidually. The binary core complex was changed as little as
possible, being an experimentally determined structure and
thus the most reliable part of the model, but some minor
changes in side chain orientation could not be avoided for
proper adaptation. An important point was to keep the
C-terminal domains of the receptor chains close together, as
this was expected to be essential for dimer formation and
thereby signaling through the membrane. The structures of
c
c
and the ternary complex were modeled so that residues
that exhibit positive coupling energies during double-
mutant cycle analysis were placed close to each other.
Occasionally, however, there was a Ôconflict of interestÕ
between the requirements of interaction and those of
dimerization.
DISCUSSION
This mutational analysis defines human c
c
residues involved
in IL-4 binding. The residues are located in the EF1, BC2,
and FG2 loops and the interdomain segment of c
c
.The
functional binding epitope of c
c
includes residues I100,
L102, Y103, and L208 as major binding determinants and
five residues, N128, H159, L161, E162, and G210, as minor
determinants. Our results also show that the truncated c
c
CHR has the same binding affinity as the complete c
c
ectodomain, indicating that the short N-terminal region of
c
c
is not required for ligand binding. This is true for most
type I cytokine receptors, except for hgp130 [51] and
granulocyte colony-stimulating factor receptor [54]. There-
fore, c
c
CHR, the short form of the c
c
ectodomain, may be
Table 2. Double mutant cycle analysis of interaction between c
c
and
IL-4. The dissociation constants K
d
were evaluated from equilibrium
binding between wild-type (wt) or mutants (mut) of the c
c
ectodomain
and immobilized IL-4BP saturated with wild-type or mutants of IL-4.
The loss of free energy of binding on mutation was calculated as
ddG ¼5.69 log K
d
(mut)/K
d
(wt). ddG
sum
is the sum of the losses of free
energy of binding upon mutation for IL-4 and c
c
separately. ND,
Sensogram could not be evaluated because of weak binding.
IL-4
variants
c
c
chain
variants
K
d
(l
M
)
ddG
(kJÆmol
)1
)
ddG
sum
(kJÆmol
)1
)
wt wt 4.0
N15A wt 20 4.0
R121A wt 12 2.8
Y124F wt 6.9 1.4
S125A wt 8.5 1.9
wt N128A 15 3.2
wt H159A 17 3.5
wt L161A 13 2.9
wt E162A 13 2.9
wt G210A 15 3.2
N15A N128A 34 5.3 7.2
N15A H159A 61 6.7 7.5
N15A L161A 75 7.2 6.9
N15A E162A ND – 6.9
N15A G210A 59 6.7 7.2
R121A N128A 50 6.2 6.0
R121A H159A 75 7.2 6.3
R121A L161A 27 4.7 5.7
R121A E162A 127 8.6 5.7
R121A G210A 81 7.4 6.0
Y124F N128A 22 4.2 4.6
Y124F H159A 33 5.2 4.9
Y124F L161A 24 4.5 4.3
Y124F E162A 83 7.5 4.3
Y124F G210A 29 4.9 4.6
S125A N128A 19 3.9 5.1
S125A H159A 57 6.6 5.4
S125A L161A 53 6.4 4.8
S125A E162A 37 5.5 4.8
S125A G210A 22 4.2 5.1
Table 3. Co-operativity between residue pairs in the interaction interface of c
c
and IL-4. The coupling energy between a pair of residues was calculated
as ddG
int
¼ddG
sum
)ddG (data from Table 2) according to eqn (1). The underlined values indicate favorable interaction. The numbers in
parentheses are the calculated errors (2 r,a¼0.95). ND, Sensogram could not be evaluated because of weak binding.
c
c
chain
variants
ddG of IL-4 variants (kJÆmol
)1
)
N15A R121A Y124F S125A
N128A 1.9 (0.62) )0.2 (0.66) 0.4 (0.80) 1.2 (0.90)
H159A 0.8 (0.64) )0.9 (1.10) )0.3 (0.83) )1.2 (0.86)
L161A )0.3 (0.99) 1.0 (0.80) )0.2 (0.64) )1.6 (0.72)
E162A ND )2.9 (0.90) )3.2 (0.76) )0.7 (0.68)
G210A 0.4 (0.66) )1.4 (0.87) )0.3 (0.48) 0.9 (0.67)
1494 J.-L. Zhang et al.(Eur. J. Biochem. 269)ÓFEBS 2002


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)