
The crystal structure of glucose-6-phosphate isomerase from
Leishmania mexicana
reveals novel active site features
Artur T. Cordeiro
1
, Paul A. M. Michels
2
, Luiz F. Delboni
3
and Ota
´vio H. Thiemann
1
1
Laboratory of Protein Crystallography and Structural Biology, Physics Institute of Sa
˜o Carlos, University of Sa
˜o Paulo,
Sa
˜o Carlos-SP, Brazil;
2
Research Unit for Tropical Diseases and Laboratory of Biochemistry, Christian de Duve Institute of
Cellular Pathology, Brussels, Belgium;
3
Pontificia Universidade Cato
´lica de Minas Gerais, Poc¸ os de Caldas-MG, Brazil
Glucose-6-phosphate isomerase catalyzes the reversible
aldose-ketose isomerization of
D
-glucose-6-phosphate to
D
-fructose-6-phosphate in glycolysis and gluconeogenesis,
and in the recycling of hexose-6-phosphate in the pentose
phosphate pathway. The unicellular protozoans, Trypan-
osoma brucei,T. cruzi and Leishmania spp., of the order
Kinetoplastida are important human parasites responsible
for African sleeping sickness, Chagas’ disease and leish-
maniases, respectively. In these parasites, glycolysis is an
important (and in some cases the only) metabolic pathway
for ATP supply. The first seven of the 10 enzymes that
participate in glycolysis, as well as an important fraction
of the enzymes of the pentose phosphate pathway, are
compartmentalized in peroxisome-like organelles called
glycosomes. The dependence of the parasites on glycolysis,
the importance of the pentose phosphate pathway in
defense against oxidative stress, and the unique compart-
mentalization of these pathways, point to the enzymes
contained in the glycosome as potential targets for drug
design. The present report describes the first crystallo-
graphic structure of a parasite (Leishmania mexicana)
glucose-6-phosphate isomerase. A comparison of the
atomic structure of L. mexicana, human and other
mammalian PGIs, which highlights unique features of the
parasite’s enzyme, is presented.
Keywords:Leishmania; phosphoglucose isomerase; glycoly-
sis; substrate–enzyme; human PGI.
Leishmania mexicana is a human protozoan pathogen
belonging to the order Kinetoplastida [1,2]. Among the
kinetoplastid organisms, several human parasites are pre-
sent, including Trypanosoma brucei,T. cruzi and various
Leishmania species that are responsible for diseases such as
African sleeping sickness, Chagas’ disease and leishmani-
ases, respectively, causing serious health problems in
tropical and subtropical areas, which, in several cases, are
fatal if left untreated. This scenario is aggravated by a lack
of effective, available drugs for the treatment of infected
individuals, and the reports of drug-resistant parasite
strains. Leishmania infection may lead to disorders that
can manifest themselves in three different clinical forms –
cutaneous, visceral and mucocutaneous leishmaniasis –
depending on the Leishmania species involved. The actual
treatment for leishmaniasis is based mainly on antimonial
compounds that are of low specificity and cause undesirable
side-effects [1,2].
Glycolysis is an important, and in some cases the only,
metabolic pathway for the ATP supply of these parasites.
The first seven of the 10 enzymes that participate in
glycolysis are compartmentalized in peroxisome-like organ-
elles called glycosomes [3], a characteristic of all members of
the Kinetoplastida order. A consequence of this organellar
localization is that the kinetoplastid glycolytic enzymes
differ in many kinetic and structural properties from their
counterparts in other organisms, and that the flux through
the pathway is regulated in a different manner [2,3]. Not
only glycolysis is found in glycosomes; also found is a
significant fraction of many enzymes of the pentose
phosphate pathway [4,5], which uses sugars for the forma-
tion of
D
-ribose-5-phosphate for nucleotide synthesis and
NADPH, for biosynthetic processes, and for defense against
oxidant stress. This process is also very important for the
trypanosomes and leishmanias, particularly to combat
oxidative attack by the host. Therefore, both the glycolytic
and pentose phosphate pathways have been indicated as
promising drug targets [2,6,7].
Glucose-6-phosphate isomerase (often still called by its
old name, phosphoglucose isomerase; PGI) is the second
enzyme in glycolysis and catalyzes the reversible aldose-
ketose isomerization of
D
-glucose 6-phosphate (
D
-Glc6P)to
D
-fructose 6-phosphate (
D
-Fru6P). It is also an enzymatic
link between glycolysis and the pentose phosphate pathway.
Correspondence to O. H. Thiemann, Laboratory of Protein Crystal-
lography and Structural Biology, Department of Physics and Infor-
matics, Physics Institute of Sa
˜o Carlos, University of Sa
˜oPaulo,
Avenue Trabalhador Sa
˜ocarlense 400, PO Box 369, 13566–590, Sa
˜o
Carlos-SP, Brazil. Fax: + 55 16 273 9881, Tel.: + 55 16 273 8089,
E-mail: thiemann@if.sc.usp.br
Abbreviations:
D
-Fru6P,
D
-fructose-6-phosphate;
D
-Glc6P,
D
-glucose-
6-phosphate; dPGI-Lm, N-terminally deleted glucose-6-phosphate
isomerase from Leishmania mexicana; PGI, glucose-6-phosphate
isomerase; PGI-Lm, glucose-6-phosphate isomerase from
Leishmania mexicana.
Enzyme: glucose-6-phosphate isomerase (E.C. 5.3.1.9).
Note: The PDB ID code for the solution structures of
Leishmania mexicana glucose-6-phosphate isomerase full-length
PGI-Lm is 1Q5O and of the form with the 48 residues deleted from
its N-terminus (dPGI-Lm), 1Q1I.
(Received 3 December 2003, revised 31 March 2004,
accepted 6 May 2004)
Eur. J. Biochem. 271, 2765–2772 (2004) ÓFEBS 2004 doi:10.1111/j.1432-1033.2004.04205.x

In the pentose phosphate pathway, PGI recycles one of the
products (
D
-Fru6P) back into the substrate (
D
-Glc6P)for
glucose-6-phosphate dehydrogenase, representing the initial
step of the pathway.
The pentose phosphate pathway appears to have a dual
localization in both Leishmania spp. and T. brucei,as
several of its enzymes have been shown to be present in both
the cytosol and the glycosomes [4,5]. Whereas most
glycolytic enzymes are entirely or predominantly present
inside the organelles, PGI, the enzyme shared with the
pentose phosphate pathway, is indeed found in both
compartments, although in a ratio that differs between
Kinetoplastida species. In bloodstream-form T. brucei,
most (85%) PGI resides in the glycosomes, but in
cultured L. mexicana promastigotes (representative of the
insect-infective stage), PGI activity was detected mainly in
the cytosol, with the remainder (less than 10%) associated
with the glycosome [8,9]. This is consistent with a higher
pentose phosphate pathway activity and a lower glycolytic
activity in promastigotes when compared to the blood-
stream form of T. brucei [10–12]. The involvement of PGI in
two important pathways of Leishmania metabolism may
make it interesting for drug targeting. Recently, a 50%
growth inhibition in bloodstream-form T. brucei was
observed as a consequence of decreasing the level of PGI
by RNA interference. This result is indicative of the central
role of PGI in the parasite metabolism [13].
In this report we present the atomic structure of
L. mexicana PGI (PGI-Lm) obtained by X-ray diffraction
techniques. The comparison of this structure with the
available mammalian PGI structures allowed the identifi-
cation of significant differences between the enzyme of the
parasite and its human homologue, which may be exploited
in future drug design.
Experimental procedures
Crystallization data collection and structure
determination
Two different constructs of the PGI gene from L. mexicana
have been expressed in Escherichia coli BL21(DE3) and
purified to homogeneity [14]. The two forms correspond to
the entire 604 amino acid PGI sequence (PGI-Lm) and a
polypeptide from which the N-terminal 47 amino acid
residues were deleted (dPGI-Lm), respectively. The
N-terminal deletion from L. mexicana PGI does not
interfere with the catalytic process. The N-terminal exten-
sion is believed to represent an unorganized structure,
possibly related to the glycosomal localization of the protein
[8,9,14], and could be interfering in the crystallization
process. Both forms of the bacterially expressed L. mexica-
na PGI (PGI-Lm and dPGI-Lm) were successfully crystal-
lized by the hanging drop vapor-diffusion technique, but the
dPGI-Lm crystals presented a better intrinsic order [14].
A complete PGI-Lm dataset of 228 frames was collected
at 100 K from a single crystal grown under conditions
previously reported [14], using a RIGAKU X-ray source
and a MAR345 image plate detector. The crystal-
to-detector distance was set to 250 mm and each frame
was exposed for 4 min with a phi oscillation of 0.75°.The
dataset was processed using
DENZO
and
SCALEPACK
[15].
The molecular replacement solution obtained in the
previous work was used as an initial model in the
refinement procedure. Crystals of dPGI-Lm were soaked
for 5 min in cryoprotectant solution consisting of the
reservoir solution and 17.5% methane pentanediol (w/v)
containing 3.0 m
MD
-Fru6Pand then flash-frozen in liquid
nitrogen. A complete dataset of 140 frames was collected
at 100 K for the dPGI-Lm at the X25 beam line of the
Brookhaven National Laboratory. This beam line is
equipped with a Q315 ccd detector, which allowed the
collection of reflections at a 2.3 A
˚resolution limit with the
detector placed 325 mm from the crystal. Each frame was
exposed for 15 s with a phi oscillation of 0.75°.Theframes
were integrated using
MOSFLM
[16] and the reflections were
scaled using
SCALA
from the CCP4 package [17]. The
refined PGI-Lm structure, collected using the RIGAKU
X-ray source, was used as initial model in the refinement
of the dPGI-Lm.
Structure refinement
All refinement procedures were performed using the
CNS
program [18], except for a final TLS parameters refine-
ment performed with
REFMAC
5 from the CCP4 package
[17]. The first structure refined was the PGI-Lm. One
monomer of the previously reported solution, obtained
using the
AMORE
program [19], was submitted to a rigid
body routine with data ranging between 30 and 3 A
˚
resolution. Several cycles of simulated annealing, using the
maximum likelihood method, were performed, followed
by coordinate and B-factor refinement using all data up to
the 2.6 A
˚resolution border. Local corrections and
N-terminal amino acid residue additions were per-
formed using the
O
program [20] while inspecting the
2r
A
|F
o
|-D|F
c
|mapandthem|F
o
|-D|F
c
|differencemap.
Water molecules were introduced running the Ôwater-pickÕ
script of
CNS
. Finally, all model atoms were used to define
a single group for refinement of TLS parameters with
REFMAC
5 [17].
The dPGI-Lm data were refined following the same
procedures adopted for the PGI-Lm. The water molecules
and additional N-terminal residues from the refined PGI-
Lm structure were removed from dPGI-Lm. The modified
dPGI-Lm polypeptide chain was used as an initial model for
the rigid body refinement. Prior to water addition,
D
-Fru6P
was placed into the only large electron density cloud
observed at the difference map contoured at 5 s. The correct
orientation of the phosphate group, relative to the protein
residues involved in its coordination, was driven by
structural similarity to the previously reported rabbit PGI
in complex with
D
-Fru6P[21]. Additional information of
the
D
-Fru6Pconformation was obtained by observing the
2r
A
|F
o
|-D|F
c
| map contoured at 1 s. Water molecules were
added using the Ôwater-pickÕscript of
CNS
with the same
parameters as applied to the PGI-Lm. A single group
containing all atoms from the model was used for refine-
ment of TLS parameters with
REFMAC
5[17].
Structure comparison
Superposition and root mean square (r.m.s.) calculations
were performed using the Caatoms and the
SWISS PDB
2766 A. T. Cordeiro et al.(Eur. J. Biochem. 271)ÓFEBS 2004

VIEWER
program [22].
D
-Fru6Pand dPGI-Lm side-chain
contacts were analyzed by using the
LIGPLOT
program [23].
Figures were prepared using the
PYMOL
program [24].
Results and discussion
Structure refinement
The previously reported molecular replacement solution
[14] was characterized as containing one homodimer
molecule per asymmetric unit in the P61 space group.
The processing of the data described in this study indicate
that the noncrystallographic twofold axe, which relates the
monomers in the asymmetric unit, described previously, is
coincident to a real crystal axe from the higher-symmetry
P6122 space group. The choice of P6122 crystal space
group reduced the asymmetric unit content to a single
monomer. The first Leishmania PGI structure refined,
PGI-Lm (pdb code, 1Q5O), had a total of 216 water
molecules added, resulting in an R
factor
value of 19.5%
andanR
free
value of 24.9%. The refinement of dPGI-
Lm-
D
-Fru6Pwas concluded with R
factor
and R
free
values
of 22.0 and 25.9%, respectively. A total of 180 water
molecules and one
D
-Fru6Pare present in the final dPGI-
Lm/
D
-Fru6Pmodel (pdb code, 1Q1I). The refinement of
TLS parameters contributed to a decrease of 2% in final
RandR
free
values for both molecules. Additional
information of both refined structures is presented in
Table 1.
Description of structures
The L. mexicana PGI structure is a homodimer and the
monomer subunit is composed of a large and a small a/b
sandwich domain and has an extended C-terminal
segment (comprising two a-helices) that embraces the
other monomer (Fig. 1). There are two catalytic sites per
dimer molecule; the catalytic sites are located in the dimer
interface formed by adjacent monomers. The PGI-Lm has
an overall fold similar to those described for rabbit,
human and pig PGIs [25–27]. The main difference is the
presence of an N-terminal sequence in PGI-Lm, which
may be involved in the functioning of the enzyme inside
the glycosome, as discussed previously [8,9,14]. For the
first 44 and the last residue – Leu605 – of PGI-Lm
(Fig. 2), no electron density was distinguishable in the
2r
A
|F
o
|-D|F
c
| map of both chains of the homodimer. The
absence of continuous electron density for residues 1–44
indicates that the N-terminal sequence is not ordered.
Based on the electron density map and sequence align-
ment, four additional residues (Val45 to Ser48) could be
added to that region of PGI-Lm (Fig. 2). The super-
position of the PGI-Lm and the N-terminally deleted
dPGI-Lmresultedinanr.m.s.of0.4A
˚.Themain
difference between the two structures is found in a
10-residue loop (of amino acids Gly433 to Ala442) in
which the catalytic His441 is located. Owing to the
improved electron density map and higher-resolution data
obtained with dPGI-Lm crystals, it was possible to
identify the correct Catrace for this 10-residue loop.
Differences vs. mammalian PGIs
A significant difference in Ca-r.m.s. can be observed in part
of the small domain of Leishmania PGI compared with its
Table 1. Statistics for data collection and refinement. dPGI-Lm,
N-terminally deleted glucose-6-phosphate isomerase from Leishmania
mexicana; PGI-Lm, glucose-6-phosphate isomerase from Leishma-
nia mexicana.
Structure PGI-Lm dPGI-Lm
Data collection
Space group P6
1
22 P6
1
22
Cell dimension
a, b and c (A
˚) 85.74, 85.74
and 350.43
85.13, 85.13
and 350.09
a,band c(°) 90, 90 and 120 90, 90 and 120
Resolution range (A
˚) 30–2.6 74.53–2.35
Unique reflections 24333 32626
Redundancy 6.7 7.1
Completeness (last shell) (%) 98.5 (98.8) 100 (100)
R-sym (%) 5.8 6.1
Refinement
Number of atoms
Protein 8607 8649
Heteroatoms 0 32
Solvent 216 180
Used reflections 23091 30902
R-factor 19.5 22.0
R-free 24.9 25.9
Rms
bond 0.022 0.018
angle 1.83 1.69
<B-factor> (A
˚
2
) 30.9 32.8
Fig. 1. Cartoon representation of Leishmania mexicana glucose-6-
phosphate isomerase (PGI-Lm). Thenativeenzymeisahomodimer
with the monomer subunit formed by two a/bsandwich domains
(large and small domains) and a C-terminal a-helix segment that
embraces the adjacent monomer. The catalytic residues are distributed
among the small domain and C-terminal segment from one monomer,
and at the large domain from the other monomer.
ÓFEBS 2004 Leishmania PGI crystal structure (Eur. J. Biochem. 271) 2767

human homologue [28] (pdb code, 1jlh), although both
present the same overall fold. The small domain from all
PGIs is connected to the large domain by two long a-helices
named LH-n (long helix connected to the small domain N-
terminus) and LH-c (long helix connected to the small
domain C-terminus). The PGI-Lm small domain encom-
passes residues 197–316. The superposition of Leishmania
and human PGI (pdb code, 1jlh), using the large domain
and the first a-helix (h-l) of the small domain (Fig. 2), results
in a Ca-r.m.s. of 0.9 A
˚for the considered residues. The
calculated Ca-r.m.s. for the remaining residues (small
domain, except for the h-l a-helix) is 3.3 A
˚(Fig. 3B).
Superposition of just the small-domain residues, including
the h-l a-helix, from each monomer results in a mean Ca-
r.m.s. of 0.7 A
˚. This r.m.s. analysis indicates a rigid body
displacement between the large and small domains when
comparing human and Leishmania PGI.
The fact that dimer assembly is driven by contacts
between residues located exclusively in the large domain
(Fig. 1) suggests that residues connecting large and small
domains from the same monomer are responsible for the
differences between the PGI structures. Moreover, the high
Fig. 2. Structure-based alignment of glucose-6-phosphate isomerase (PGI) sequences from different organisms: Leishmania mexicana,
Trypanosoma brucei (Tryp; SwissProt code: P13377), human [28], rabbit [25] and pig [27]. The secondary elements of N-terminally deleted glucose-6-
phosphate isomerase from Leishmania mexicana (PGI-Lm) are represented as h (a-helices) and s (b-sheet strands). The small-domain helix and sheet
elements are underlined. Residues in bold are involved in the coordination of the substrate’s phosphate group. Positions of the possible catalytic
residues, described in previous work [21], are colored in magenta. In the Leishmania (Leish) sequence, the residues shown in italics are not seen in the
electronic density maps of PGI-Lm; the PGI-Lm residues in the black box have a mean Car.m.s. deviation of 3.3 A
˚whensuperimposedtothe
human PGI [28]. The PGI-Lm residues in the gray box superimpose with human PGI with a mean Car.m.s. of 0.9 A
˚. Loops A and B are colored
green and yellow, respectively. PGI-Lm residues, marked above the alignment using the letter ÔcÕ, are in contact with residues of the opposite
monomer by a distance of less than 3.6 A
˚.TheÔoÕmarks residues of PGI-Lm that are in hydrophobic contact with Met337 (marked with #).
2768 A. T. Cordeiro et al.(Eur. J. Biochem. 271)ÓFEBS 2004
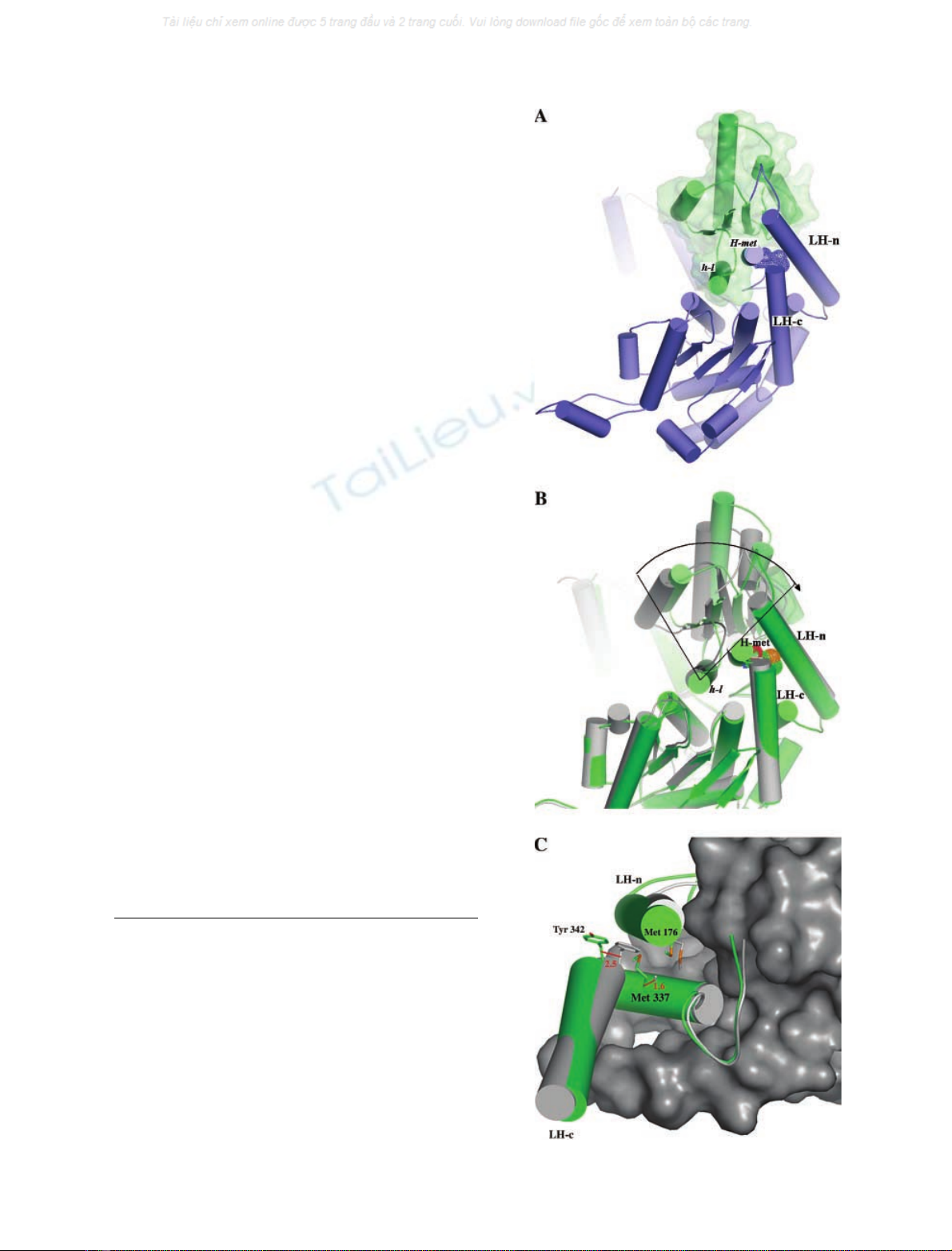
structural similarity of the small domain region (r.m.s. of
0.7 A
˚), observed between the mammalian and parasite
PGIs, point to a conserved packing of the secondary
structure elements of this domain. Amino acid substitutions
in the small domain do not result in significant alterations of
the domain interfaces between the two PGIs. We identified,
from this superposition analysis, that the presence of
Met337 in PGI-Lm, and Ala284 at a structurally equivalent
site in the human PGI, contribute significantly to the small-
domain position differences observed between both struc-
tures. Met337 is located in an a-helical segment connecting
the C-terminal side of the small domain to the LH-c of the
large domain (Fig. 2).
The first a-helix of the small domain (h-l) is positioned
adjacent to the large domain, establishing the main inter-
domain contacts (Fig. 3A). The mean Ca-r.m.s. for h-l,
calculated between human and PGI-Lm, is equivalent to the
Ca-r.m.s. calculated between the large domains (< 0.9 A
˚).
The direction of the h-l a-helix can be associated with an
imaginary axis describing a rigid body movement of the
small domain between the superimposed structures
(Fig. 3B). The substitution of Ala284 in human PGI with
Met337 in PGI-Lm causes a local stress in both long a-
helices (LH-n and LH-c) connecting the large and small
domains. Met337 is located at a short a-helix segment (H-
met), side-by-side with h-l of the small domain, and makes
hydrophobic contacts to conserved residues in LH-c and
LH-n. The Caatoms of Met337 in PGI-Lm and Ala284 in
human PGI are separated by a distance of 1.6 A
˚, while the
Caatoms of the first residue of LH-n, Tyr342 (in PGI-Lm)
and Phe289 (in human PGI) are at a distance of 2.5 A
˚
(Fig. 3C). Finally, it is clear from the structural superpo-
sition that PGI-Lm presents its small domain in a more
open conformation when compared to the human homo-
logue, resulting in a larger active-site cavity. This may
explain the difference in the affinity of the proteins for their
substrate and product. A higher K
m
value for the substrate
D
-Fru6Pwas measured for PGI-Lm (242 l
M
) when com-
pared with that of the human enzyme (99 l
M
) [14].
Substrate binding
The position of
D
-Fru6Pin the dPGI-Lm structure is clearly
seen in the difference map calculated in the absence of the
ligand (Fig. 4A). However, as it is not clear whether
D
-7Fru6Pis in the open or closed conformation, both
Fig. 3. Structural details of the Leishmania PGI. (A) Cartoon repre-
sentation of large (blue) and small (green) domains from Leishmania
mexicana glucose-6-phosphate isomerase (PGI-Lm). The large and
small domains are connected by two long a-helices, named LH-n and
LH-c; Met337 is located at a-helix ÔH-metÕin the large domain, which is
placed side-by-side with Ôh-lÕin the small domain (B). Superimposed
monomers from human (gray) [28] and PGI-Lm (green) highlight the 2
A
˚side-displacement of their small domains. An imaginary rotation axis
can be placed close to h-l, indicated by the vertices of the drawn lines.
(C) Detailed view of Met337 neighboring residues located in both LH-n
and LH-c of PGI-Lm (green). The small domain from human PGI is
represented by the gray surface. The presence of Met337 at the cor-
responding position of Ala284 in human PGI causes the displacement
of Tyr342 (Phe286 in human PGI) located in the LH-c of PGI-Lm.
ÓFEBS 2004 Leishmania PGI crystal structure (Eur. J. Biochem. 271) 2769


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)