
Kinetic mechanism for p38 MAP kinase a
A partial rapid-equilibrium random-order ternary-complex
mechanism for the phosphorylation of a protein substrate
Anna E. Szafranska
1
and Kevin N. Dalby
1,2
1 Division of Medicinal Chemistry, University of Texas at Austin, TX, USA
2 Graduate Programs in Biochemistry and Molecular Biology and the Center for Molecular and Cellular Toxicology, University of Texas at
Austin, TX, USA
Keywords
docking; inhibition; kinetic mechanism;
MAP kinase; p38 MAPK
Correspondence
K. N. Dalby, Division of Medicinal
Chemistry, College of Pharmacy, University
of Texas at Austin, TX 78712, USA
Fax: +1 512 232 2606
Tel: +1 512 471 9267
E-mail: Dalby@mail.utexas.edu
(Received 28 February 2005, revised
18 May 2005, accepted 20 June 2005)
doi:10.1111/j.1742-4658.2005.04827.x
p38 Mitogen-activated protein kinase alpha (p38 MAPKa) is a member of
the MAPK family. It is activated by cellular stresses and has a number
of cellular substrates whose coordinated regulation mediates inflammatory
responses. In addition, it is a useful anti-inflammatory drug target that has
a high specificity for Ser-Pro or Thr-Pro motifs in proteins and contains a
number of transcription factors as well as protein kinases in its catalog
of known substrates. Fundamental to signal transduction research is the
understanding of the kinetic mechanisms of protein kinases and other pro-
tein modifying enzymes. To achieve this end, because peptides often make
only a subset of the full range of interactions made by proteins, protein
substrates must be utilized to fully elucidate kinetic mechanisms. We show
using an untagged highly active form of p38 MAPKa, expressed and puri-
fied from Escherichia coli [Szafranska AE, Luo X & Dalby KN (2005) Anal
Biochem 336, 1–10) that at pH 7.5, 10 mmMg
2+
and 27 C p38 MAPKa
phosphorylates ATF2D115 through a partial rapid-equilibrium random-
order ternary-complex mechanism. This mechanism is supported by a
combination of steady-state substrate and inhibition kinetics, as well as
microcalorimetry and published structural studies. The steady-state kinetic
experiments suggest that magnesium adenosine triphosphate (MgATP),
adenylyl (b,c-methylene) diphosphonic acid (MgAMP-PCP) and magnes-
ium adenosine diphosphate (MgADP) bind p38 MAPKawith dissociation
constants of K
A
¼360 lm,K
I
¼240 lm, and K
I
> 2000 lm, respectively.
Calorimetry experiments suggest that MgAMP-PCP and MgADP bind
the p38 MAPKa–ATF2D115 binary complex slightly more tightly than
they do the free enzyme, with a dissociation constant of K
d
70 lm.
Interestingly, MgAMP-PCP exhibits a mixed inhibition pattern with
respect to ATF2D115, whereas MgADP exhibits an uncompetitive-like
pattern. This discrepancy occurs because MgADP, unlike MgAMP-PCP,
binds the free enzyme weakly. Intriguingly, no inhibition by 2 mmaden-
ine or 2 mmMgAMP was detected, suggesting that the presence of a
b-phosphate is essential for significant binding of an ATP analog to the
Abbreviations
ATF2D115, glutathione S-transferase fusion protein of activating transcription factor 2 residues 1–115; ERK, extracellular signal-regulated
kinase; ITC, isothermal titration calorimetry; JNK, Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MgADP, magnesium
adenosine diphosphate; MgAMP-PCP, adenylyl (beta,gamma-methylene) diphosphonic acid; MgATP, magnesium adenosine triphosphate;
MKK3, MAP kinase kinase 3; MKK6, MAP kinase kinase 6; MKP3, MAP kinase phosphatase; NADH, nicotinamide adenine dinucleotide;
p38 MAPKa, p38 mitogen-activated protein kinase alpha.
FEBS Journal 272 (2005) 4631–4645 ª2005 FEBS 4631

All organisms, from bacteria and yeasts to mammalian
cells, respond to stimuli from the extracellular environ-
ment. Incoming signals are sent via a cascade of pro-
teins and enzymes from the surface of cells to their
interior, causing alterations in gene expression and
protein activity. These, in turn, generate cellular
responses, such as growth, differentiation, inflamma-
tion and apoptosis. In eukaryotic cells, the mitogen-
activated protein kinase (MAPK) module is a key
element in the propagation, amplification and trans-
port of extracellular signals to the nucleus [1]. The
MAPK superfamily includes the extracellular signal-
regulated kinases (ERKs), the Jun N-terminal kinases
(JNKs) and the p38 MAP kinases, among others.
These enzymes are terminal components of three-tiered
MAPK modules, each of which consists of a MAP
kinase (MAPK), a MAPK kinase (MAPKK) and a
MAPKK kinase (MAPKKK). MAPK modules oper-
ate in numerous biological settings where, through
largely unknown mechanisms, multiple components
impinge on a particular MAPKKK [1].
In recent years there has been substantial interest
in MAPKs due to their participation in numerous bio-
logical pathways and various human conditions and
diseases. One notable MAPK is p38 MAPKawhose
activity has been associated with diseases such as can-
cer [2] or those with inflammatory components [3–5].
p38 MAPKais phosphorylated on Tyr180 and Thr182
by the upstream activators MAP kinase kinase 3
(MKK3) and MAP kinase kinase 6 (MKK6). Once
activated, p38 MAPKaexerts its effect by directly
phosphorylating transcription factors such as activa-
ting transcription factor 2 (ATF2) and MEF2, or indi-
rectly by activating downstream protein kinases such
as MAPKAP-K2 and MAPKAP-K3, which in turn
phosphorylate their own substrates [1].
Despite a wealth of biological information, there are
many unsolved issues concerning this and other
MAPK signaling cascades. Within the past decade,
four isoforms of p38 MAPK termed a,b,cand dhave
been discovered, whose precise biological roles remain
to be defined [1]. Notably, the aand bisoforms are
inhibited by the classic family of pyridinyl inhibitors
related to SB 203580, whereas the cand disoforms are
not. Thus, use of SB 203580, which has been the main
pharmacological tool employed to date, is transparent
to two of the p38 MAPK isoforms. Although a num-
ber of structural studies have been reported, showing
for example, inactive p38 MAPKawith and without
inhibitors bound at the ATP site [6–12], the structure
of an enzyme–substrate complex is notably lacking.
Although a number of mutagenesis studies have
mapped sites of protein–protein interaction, the basis
for and extent of the differences in specificity within
the p38 MAPK family are still poorly understood.
Thus, we have no clear picture of how p38 MAPKs
recognize protein substrates, or how this recognition is
regulated in vivo. Furthermore, we do not know how
cellular proteins such as scaffold proteins interact with
p38 MAPK isoforms, how these interactions are regu-
lated, how they interplay with catalysis, how they may
be exploited therapeutically or how they differ within
the family.
There is currently a lot of interest in understand-
ing the molecular recognition events associated with
MAPKs, because docking domains are thought to play
a major role in determining the specificity of sub-
strate–ligand and protein–ligand interactions [13–15].
A growing number of enzymes are thought to utilize
docking domains, which are substrate recognition ele-
ments lying outside the active site of the enzyme and
which govern the formation of an enzyme–substrate
complex [16–23]. Several years ago, we showed that
despite the presence of docking domains on p38
MAPKa, which could tether a protein substrate and
facilitate multiple phosphorylations in one collision,
p38 MAPKaphosphorylates ATF2D115 on Thr69 and
Thr71 in a nonprocessive manner [24]. Prior to this
study, LoGrasso et al. reported that p38 MAPKa
phosphorylates ATF2D115 via a compulsory-order
ternary-complex mechanism, in which the binding of
ATF2D115 must precede that of magnesium ATP
(MgATP) (Scheme 1B) [25]. This possibility is intrigu-
ing because: (a) the proposed mechanism would appear
to require novel communication between the enzyme
and substrates to ensure that p38 MAPKaexclusively
binds ATF2D115 before MgATP; and (b) such proper-
ties might be due to the employment of docking
domains in substrate recognition. However, the propo-
sal of LoGrasso et al. was challenged in a report that
enzyme. Surprisingly, we found that inhibition by the well-known
p38 MAPKainhibitor SB 203580 does not follow classical linear inhibi-
tion kinetics at concentrations > 100 nm, as previously suggested, demon-
strating that caution must be used when interpreting kinetic experiments
using this inhibitor.
Kinetic mechanism for p38 MAP kinase aA. E. Szafranska and K. N. Dalby
4632 FEBS Journal 272 (2005) 4631–4645 ª2005 FEBS

asserted that p38 MAPKamust bind MgATP before
it binds a peptide substrate (Scheme 1C) [26].
Recently, we established a new protocol for the pre-
paration of recombinant murine p38 MAPKa[27],
whose activity towards ATF2D115 is some 10-fold
greater than previously reported [25]. Given the avail-
ability of a highly active untagged form of p38
MAPKa, the potential novelty of its docking domain-
dependent substrate recognition, the uncertainty of it
kinetic mechanism and the interest in the development
of protein–protein interaction inhibitors, we decided to
reinvestigate its kinetic mechanism using ATF2D115
as the substrate. We describe a steady-state kinetic
investigation of untagged p38 MAPKaand report
that rather than following a compulsory-order ternary-
complex mechanism, as previously reported [25],
p38 MAPKaphosphorylates ATF2D115 via a par-
tial rapid-equilibrium random-order ternary-complex
mechanism. We also show that nucleotides such as
MgATP and particularly magnesium ADP (MgADP)
bind preferentially to the binary p38 MAPKa–
ATF2D115 complex, whereas no binding of magnes-
ium AMP (MgAMP) or adenine was detected to
any enzyme form. This study provides the basis for
the design of further structure ⁄function and tran-
sient kinetic studies aimed at defining the kinetic mech-
anism and physical properties of p38 MAPKain
detail.
Results
Steady-state kinetics
Murine p38 MAPKawas expressed in Escherichia coli,
purified and fully activated by constitutively active
MKK6b according to the method of Szafranska and
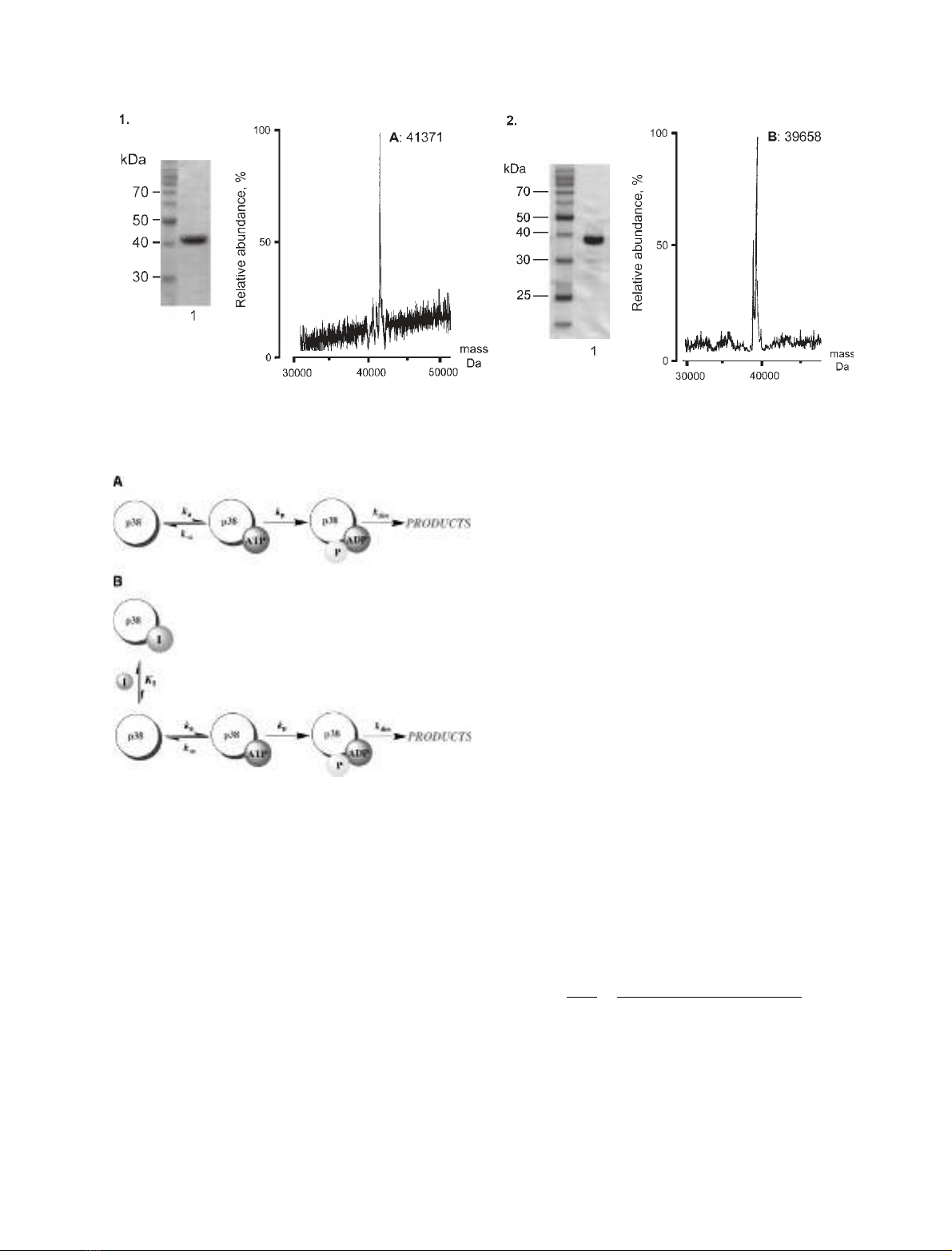
Dalby [27] (Fig. 1). This preparation corresponds to
the highest reported activity against ATF2D115 for
this enzyme [26]. To examine the propensity of
p38 MAPKato form a functional binary complex with
MgATP, the ATPase activity of the enzyme was
assessed. In line with a previous report, p38 MAPKa
displayed robust ATPase activity in the presence of
10 mmMg
2+
at pH 7.6 (k
cat
¼0.3 s
)1
and K
m
¼
353 lm) [26]. The simplest mechanism accounting for
the ATP hydrolysis is shown in Scheme 2A. According
to this mechanism, MgATP reversibly binds p38
MAPKain the active site to form the binary complex
EÆMgATP (k
a
). This binding renders it susceptible to
nucleophilic attack by hydroxyl nucleophiles, leading
to the nucleophilic addition of a water molecule to the
c-phosphoryl group of MgATP (k
p
), and the forma-
tion of MgADP and inorganic phosphate (P
i
). These
products then dissociate (k
diss
) from the active site.
Given the slow turnover (k
cat
¼0.3 s
)1
) for the hydro-
lysis reaction, and the relatively large Michaelis–
Menten constant for MgATP, we assume a rapid-equi-
librium mechanism where K
m
¼k
–a
⁄k
a
¼353 lm.A
conservative estimate for the second-order rate con-
stant of k
a
¼10
4
m
)1
Æs
)1
for the binding of MgATP to
p38 MAPKagives a rate constant for the dissociation
of MgATP from the enzyme of k
-a
¼3.5 s
)1
, if the dis-
sociation constant K
A
¼350 lmis used. This value
exceeds k
cat
by one order of magnitude, supporting the
rapid-equilibrium assumption.
The ability of p38 MAPKato bind MgATP and
facilitate the nucleophilic attack of a water molecule
with a turnover of 0.3 s
)1
, which is only fourfold lower
than the turnover of ATF2D115 (see below), supports
the notion that the EÆMgATP complex is not a dead-
end complex with respect to the binding and phos-
Scheme 1. (A) Random-order ternary-complex mechanism, (B)
compulsory-order ternary-complex mechanism (ATF2D115 binds
first, ATP second), (C) Compulsory-order ternary-complex mechan-
ism (ATP binds first, peptide second).
A. E. Szafranska and K. N. Dalby Kinetic mechanism for p38 MAP kinase a
FEBS Journal 272 (2005) 4631–4645 ª2005 FEBS 4633
phorylation of ATF2D115. Given the binding mode
adopted by peptide substrates for a number of protein
kinases, it is reasonable to assume that a protein sub-
strate can bind productively to a preformed EÆMgATP
complex. Thus, as pointed out by Chen et al. [26], the
robust ATPase activity exhibited by p38 MAPKa
sheds some doubt on the compulsory-order ternary-
complex mechanism proposed by LoGrasso et al. [25].
We expressed and purified the glutathione S-trans-
ferase (GST) fusion protein of the N-terminal 115 resi-
dues of the transcription factor ATF2 (ATF2D115)
essentially as described previously [25], with some
minor modifications (Fig. 1) [27]. Having established
the kinetic competence of the EÆMgATP complex
(with respect to nucleophilic attack by water), we con-
ducted initial rate studies at various concentrations
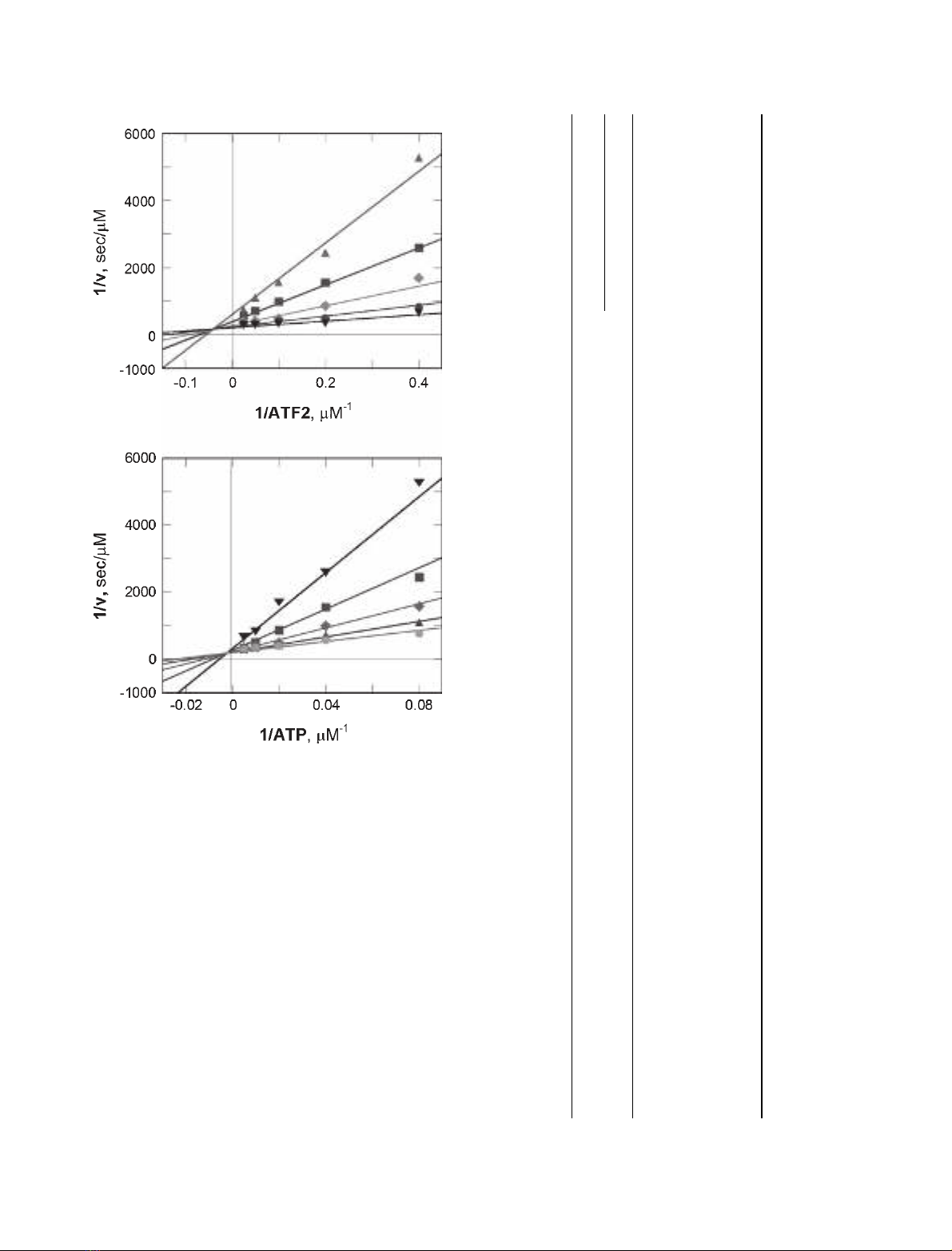
of ATF2D115 and MgATP. Reciprocal plots of initial
rate versus the concentration of ATF2D115 (Fig. 2A)
or ATP (Fig. 2B) revealed an intersecting pattern of
lines (> 1 ⁄v¼0), indicative of a sequential kinetic
mechanism, in which both substrates must bind to
form a ternary complex before catalysis occurs. Pre-
viously, we showed that ATF2D115 is phosphorylated
twice by p38 MAPKaon Thr69 and Thr71 in a non-
processive manner and that under initial rate con-
ditions, only the mono-phosphorylated forms of
ATF2D115 are produced at equal rates [24].
Our results differ in two significant aspects from
those previously reported for flag-tagged p38 MAPKa
[25]. First, in our case the double-reciprocal plots inter-
sect above the x-axis (compared with below the x-axis
for the flag-tagged enzyme). Second, the reported cata-
lytic constant towards ATF2D115 is some 10-fold
higher. It is conceivable that these differences in activ-
ity reflect the presence of an N-terminal flag tag
and ⁄or the method by which the enzymes were over-
expressed, activated and purified. In our case a sensi-
tive tryptic analysis indicates that the enzyme was fully
activated [27].
v
Vmax
¼AB
aKAKBþaKBAþaKABþAB ð1Þ
The rapid equilibrium assumption is a powerful
approach used to simplify the analysis of enzyme
mechanisms and for a ternary-complex mechanism it
provides a good approximation to the reaction path-
way when ligand-binding events are fast compared
Fig. 1. Preparation of activated p38 MAPKaand ATF2D115. (A) 10% SDS ⁄PAGE analysis showing activated, p38 MAPKa(lane 1) and its MS
analysis (M
r
41 731 Da observed; 41 726 Da calculated). (B) 12% SDS ⁄PAGE showing ATF2D115 (lane 1) and its MS analysis (M
r
39 658 Da
observed; 39 650 Da calculated).
Scheme 2. (A) Mechanism of ATP hydrolysis by p38 MAPKa.(B)
Competitive inhibition of ATP hydrolysis with EÆI dead-end com-
plex.
Kinetic mechanism for p38 MAP kinase aA. E. Szafranska and K. N. Dalby
4634 FEBS Journal 272 (2005) 4631–4645 ª2005 FEBS
with the interconversion of the central substrate and
product complexes. The lines in Fig. 2 represent the
best fit of the experimental data to Eqn (1), which
describes a rapid-equilibrium random-order ternary-
complex mechanism (Scheme 1A), according to the
parameters shown in Table 1. According to this fit,
p38 MAPKabinds both substrates in the mid micro-
molar range [K
B
¼39 lm(ATF2D115); K
A
¼360 lm
(MgATP)] to form the respective binary complexes.
We reasoned that with ligand binding to p38 MAPKa
occurring in the micromolar range and a relatively low
A
B
Fig. 2. Two-substrate dependence kinetic analysis of p38 MAPKa.
(A) Double-reciprocal plot of 1 ⁄vversus 1 ⁄ATF2D115 at five fixed
ATP concentrations (m,12lM;n,25lM;r,50lM;d,100lM;.,
200 lM). (B) Double reciprocal plot of 1 ⁄vversus 1 ⁄ATP at five
fixed ATF2D115 concentrations (.,2.5lM;n,5lM;r,10lM;m,
20 lM;d,40lM). Solid lines are the best fit through the experi-
mental data to Eqn (1).
Table 1. Kinetic constants for p38 MAPKaobtained from two-substrate steady-state kinetics, and ATPase activity and inhibition studies. C, competitive; UC, uncompetitive; M, mixed; ND,
not determined.
Activity Substrates
Substrate dependence constants (lM)
Inhibitor
Varied
substrate
Inhibition
pattern
Inhibition constants
K
A
(lM)aK
A
(lM)K
B
(lM)aK
B
(lM)k
cat
,(s
)1
)K
I
(lM)bK
I
(lM)
Kinase ATF2D115 (B),
ATP (A)
360 ± 17 13.4 ± 15 38.6 ± 7 1.4 ± 0.1 1.1 ± 0.03 ADP ATF2D115
ATP
UC
C
>2000
a
9.7 ± 0.5
AMP-PCP ATF2D115
ATP
M
C
187.4 ± 37 8.6 ± 0.5
SB 203580 ATF2D115 ND – –
ATP C 0.021 ± 0.001
b
0.021 ± 0.001
b
ATPase ATP 353 ± 52 – – – 0.3 ± 0.01 AMP-PCP ATP C 241.5 ± 13 –
a
Lower estimate.
b
K
I
,andbK
I
set equal.
A. E. Szafranska and K. N. Dalby Kinetic mechanism for p38 MAP kinase a
FEBS Journal 272 (2005) 4631–4645 ª2005 FEBS 4635


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)