
The role of the second binding loop of the cysteine protease inhibitor,
cystatin A (stefin A), in stabilizing complexes with target proteases
is exerted predominantly by Leu73
Alona Pavlova, Sergio Estrada* and Ingemar Bjo¨rk
Department of Veterinary Medical Chemistry, Swedish University of Agricultural Sciences, Uppsala Biomedical Centre, Sweden
The aim of this work was to elucidate the roles of individual
residues within the flexible second binding loop of human
cystatin A in the inhibition of cysteine proteases. Four
recombinant variants of the inhibitor, each with a single
mutation, L73G, P74G, Q76G or N77G, in the most
exposed part of this loop were generated by PCR-based site-
directed mutagenesis. The binding of these variants to
papain, cathepsin L, and cathepsin B was characterized by
equilibrium and kinetic methods. Mutation of Leu73
decreased the affinity for papain, cathepsin L and cathep-
sin B by 300-fold, >10-fold and 4000-fold, respect-
ively. Mutation of Pro74 decreased the affinity for
cathepsin B by 10-fold but minimally affected the affinity
for the other two enzymes. Mutation of Gln76 and Asn77
did not alter the affinity of cystatin A for any of the proteases
studied. The decreased affinities were caused exclusively by
increased dissociation rate constants. These results show that
the second binding loop of cystatin A plays a major role in
stabilizing the complexes with proteases by retarding their
dissociation. In contrast with cystatin B, only one amino-
acid residue of the loop, Leu73, is of principal importance for
this effect, Pro74 assisting to a minor extent only in the case
of cathepsin B binding. The contribution of the second
binding loop of cystatin A to protease binding varies with
the protease, being largest, 45% of the total binding
energy, for inhibition of cathepsin B.
Keywords: cathepsins; cystatin; cysteine proteases; papain;
second binding loop.
Cystatins are effective protein inhibitors of cysteine pro-
teases of the papain superfamily (reviewed in [1–4]). Found
both intracellularly and extracellularly, they are believed to
control the activity of normal endogenous proteases, as well
as to protect organisms from the harmful activity of
exogenous cysteine proteases [1,4–11]. They are generally
classified into three families according to their size and the
presence of internal disulfide bonds. Cystatins of family 1,
also called stefins, are small nonglycosylated proteins 11–
12 kDa in size without disulfide bonds. Family 2 cystatins
are somewhat larger, 12–14 kDa, with a structure stabi-
lized by two disulfide bonds. Kininogens, representing the
third family, are glycosylated proteins of about 50–90 kDa.
The single polypeptide chain of a kininogen contains three
domains resembling family 2 cystatins.
Cystatins competitively inhibit the activity of papain-
like cysteine proteases by binding to the active site of the
latter and forming a tight, reversible protein–protein
complex. A model of the inhibition was initially proposed
from computer docking experiments based on the X-ray
structures of papain and chicken cystatin, a family 2
member [12]. This model was later substantiated by the
X-ray structure of a complex of the family 1 cystatin,
human cystatin B (stefin B), with papain [13], the only
structure of a cystatin–protease complex determined so
far. The N-terminal segment and two hairpin loops of the
cystatin together form a hydrophobic wedge-shaped edge
that fits well into the active-site cleft of papain. The high
degree of complementarity between the interacting surfa-
ces allows the complex to form without significant
conformational changes of either papain or the inhibitor
[12–18]. Both the similar three-dimensional structures of
cystatins of families 1 and 2 [12,13,19–21] and the
pronounced sequence homology and similar fold of
cysteine proteases of the papain family [4,11,22–24]
indicate that the general aspects of the interaction model
can be extended to complexes between cystatins and other
members of this protease family. However, certain distin-
guishing features of the structures of some cysteine
proteases, such as the occluding loop of cathepsin B
[25], cause the mode of inhibition to deviate somewhat for
these enzymes. Cystatins thus inhibit cathepsin B by a
two-step reaction involving displacement of the occluding
loop of the protease in the second step [26,27]. Moreover,
it is apparent that the role of an individual binding region
Correspondence to I. Bjo
¨rk, Department of Veterinary Medical
Chemistry, Swedish University of Agricultural Sciences,
Uppsala Biomedical Centre, Box 575, SE-751 23 Uppsala, Sweden.
Fax: + 46 18 550762, Tel.: + 46 18 4714191,
E-mail: Ingemar.Bjork@vmk.slu.se
Abbreviations: app, subscript denoting an apparent equilibrium or rate
constant determined in the presence of an enzyme substrate; E-64,
4-[(2S,3S)-3-carboxyoxiran-2-carbonyl-
L
-leucylamido]butylguani-
dine; His-tag, 10 successive histidine residues fused to an expressed
protein; k
ass
, bimolecular association rate constant; K
d
, dissociation
equilibrium constant; k
diss
, dissociation rate constant; K
i
,inhibition
constant; k
obs
, observed pseudo-first-order rate constant.
*Present address: PET-Centre, Uppsala University, University
Hospital, SE-751 85 Uppsala, Sweden.
(Received 12 July 2002, revised 16 September 2002,
accepted 20 September 2002)
Eur. J. Biochem. 269, 5649–5658 (2002) FEBS 2002 doi:10.1046/j.1432-1033.2002.03273.x
of the inhibitor in protease binding can differ with the
target protease [28].
The contributions of the N-terminal region and the first
binding loop of family 1 and 2 cystatins, as well as of the
second binding loop of family 2 cystatins, to the inhibition
of cysteine proteases have been elucidated [29–36]. Recent
work has also demonstrated the importance of two amino-
acid residues, Leu73 and His75, in the second binding loop
of the family 1 inhibitor, cystatin B, for high-affinity
binding to a number of cysteine proteases [37]. The sequence
of the corresponding hairpin loop in cystatin A (stefin A),
another member of family 1, is appreciably different from
that in cystatin B; in particular, His75 of cystatin B is
substituted by Gly in cystatin A [1]. Moreover, the NMR
structure of cystatin A shows that the second loop of this
inhibitor is highly flexible, which might be expected to affect
the interactions with the protease [20]. It is thus unclear
whether the second binding loop of cystatin A fulfils the
same function as the second binding loops of cystatin B and
family 2 cystatins and also what residues of this loop in
cystatin A may participate in the interaction.
To elucidate the role of the second binding loop of human
cystatin A in the inhibition of cysteine proteases, we have
characterized the contribution of four individual amino-acid
residues within the most exposed region of this loop (from
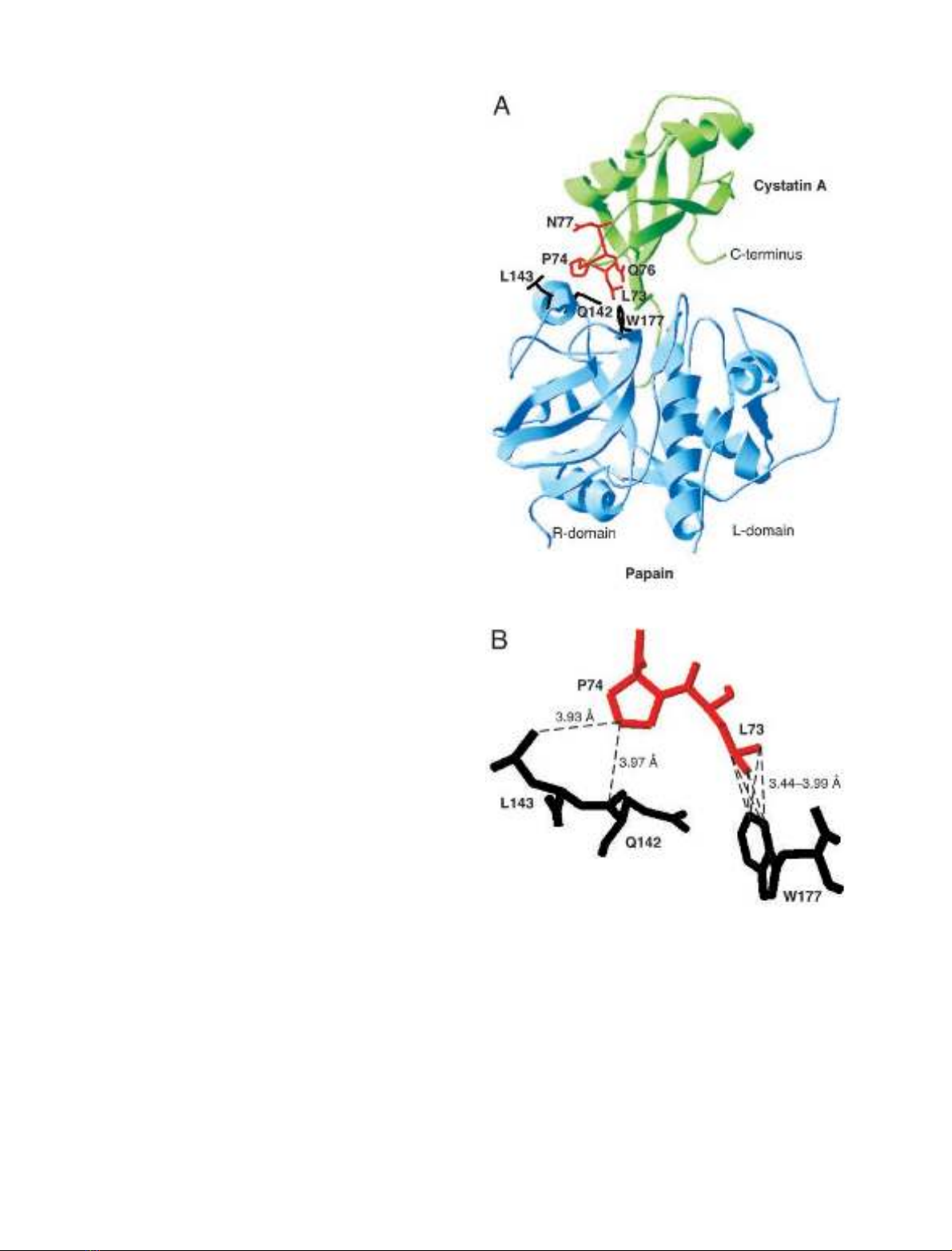
Leu73 to Asn77) to protease binding (see Fig. 1A). Four
recombinant cystatin A variants with Gly replacing each of
these amino acids were prepared, and their interaction with
papain, cathepsin L, and cathepsin B was characterized by
equilibrium and kinetic methods. The results clearly show
that the second binding loop of cystatin A is important for
the stability of complexes with cysteine proteases. Its
quantitative role in protease binding varies with the target
enzyme, but is especially important for cathepsin B. Leu73,
which is highly conserved in family 1 cystatins, makes the
predominant contribution of all residues of the loop to the
free energy of formation of the enzyme–inhibitor complex.
Pro74 is of minimal importance for the interaction with
papain and cathepsin L but participates to some extent in
cathepsin B binding. However, the roles of Gln76 and
Asn77 in the protease inhibition are negligible.
MATERIALS AND METHODS
Construction of expression vectors for cystatin A
second-loop mutants
A previously developed expression vector containing the
human cystatin A coding sequence preceded by successive
sequences for the leader peptide for the outer membrane
protein A of Escherichia coli, a His-tag, and the recognition
site for enterokinase was used in this work [38]. This vector
has a kcl857 temperature-sensitive repressor gene, allowing
induction of expression by increasing the temperature, and
an ampicillin-resistance gene [18]. Residues Leu73, Pro74,
Gln76, and Asn77 within the second binding loop of
cystatin A were substituted with Gly by PCR-based site-
directed mutagenesis [39]. Briefly, two mutagenic primers
and two standard PCR primers, the latter being comple-
mentary to regions of the vector flanking the cysta-
tin A-coding sequence, were used for creation of each
mutant (Table 1). The desired mutation was introduced in
two steps. First, two overlapping DNA fragments, bearing
Fig. 1. Model of the three-dimensional structure of the complex between
cystatin A and active papain. (A) Overall structure of the complex in
ribbon representation, with cystatin A in green and papain in blue.
Residues in the second binding loop of cystatin A mutated in this work
areinred.PapainresiduesinvolvedininteractionswiththecystatinA
second-binding-loop residues are in black. (B) Close-up view of the
interactions between residues in the second binding loop of cystatin A
and papain residues. The colors of the residues are as in (A). Inter-
molecular hydrophobic contacts within a distance of 4 A
˚are repre-
sented as dashed lines. The model is derived from the X-ray structure
of the human C3S-cystatin B–S-(carboxymethyl)papain complex
(PDB entry 1STF) [13].
5650 A. Pavlova et al.(Eur. J. Biochem. 269)FEBS 2002

the same mutation, were synthesized in two separate PCRs,
in each of which a mutagenic and a standard primer were
used and the cystatin A expression vector was the template.
In the next step, a larger DNA fragment containing the
entire mutant cystatin A-coding sequence was obtained by a
third PCR with the standard PCR primers and with a
mixture of the products of the previous two PCRs as
template. The resulting DNA fragment was cleaved with
NcoIandBamHI, and the purified cleavage product
containing the mutant cystatin A cDNA was cloned into
the original vector between the NcoIandBamHI restriction
sites, replacing the corresponding region coding for wild-
type cystatin A [38]. The vector was then transformed into
E. coli strain MC 1061, made competent with CaCl
2
[40],
and transformants were selected by growing the bacteria on
agar plates containing ampicillin. Plasmids from a number
of colonies of each mutant were purified, and those with
the correct mutant cystatin A cDNA were identified by
sequencing in an ABI PRISM310 Genetic Analyzer
(Applied Biosystems, Foster City, CA, USA).
Expression and purification of cystatin A mutants
Recombinant L73G, P74G, Q76G, and N77G cystatin A
variants were expressed in E. coli essentially as described
previously [18]. The recombinant proteins were purified
from periplasmic extracts by immobilized metal affinity
chromatography on HisBindResin (Novagen, Madison,
WI, USA), charged with Ni
2+
, or Ni/nitrilotriacetate
agarose (Qiagen, Hilden, Germany), as in previous work
[38]. The His-tag was cleaved off with enterokinase (EC
3.4.21.9; Biozyme Laboratories, Blaenavon, UK), and the
liberated cystatin A mutant was isolated by rechromato-
graphy on the same affinity column [38]. Intact His-tagged
fusion proteins still contaminating some preparations were
removed by absorption on a TALONTM Metal Affinity
Resin (Clontech, Palo Alto, CA, USA) by a hybrid batch/
gravity flow column procedure according to a protocol from
the manufacturer.
Chicken cystatin
Forms1and2ofchickencystatinwereisolatedfrom
chicken egg white [41]. The two forms have the same
sequence and are functionally identical [41], although form 2
is phosphorylated at Ser80 [42] and therefore has a lower
isoelectric point.
Proteases
Papain (EC 3.4.22.2) was purified, stored as inactive
S-(methylthio)papain and activated before use as in previ-
ous work [41]. The thiol group content of the activated
papain, determined by reaction with 5,5¢-dithiobis(2-nitro-
benzoic acid) [43], was 0.95–1.00 mol per mol of enzyme.
Titrations with chicken cystatin (form 1) [41] gave a cystatin
to papain stoichiometry of 0.98 ± 0.02, indicating that
the enzyme was fully active in binding cystatins.
Cathepsin L (EC 3.4.22.15) from sheep liver was a gift from
R. W. Mason, Alfred I. du Pont Institute, Wilmington, DE,
USA. Human liver cathepsin B (EC 3.4.22.1) was obtained
from Calbiochem (San Diego, CA, USA).
Determination of protein concentration
Most protein concentrations were calculated from
A
280
measurements. Molar absorption coefficients of
55 900
M
)1
Æcm
)1
for papain and S-(methylthio)papain
[41], 8800
M
)1
Æcm
)1
for all forms of cystatin A [18], and
11 400
M
)1
Æcm
)1
for chicken cystatin [41] were used. The
concentration of active cathepsin L was determined by
titration with 4-[(2S,3S)-3-carboxyoxiran-2-carbonyl-
L
-leu-
cylamido]butylguanidine (E-64) [44]. The concentration of
cathepsin B was provided by the manufacturer.
Binding stoichiometries
The stoichiometries of binding of the cystatin A variants to
papain were determined at least in duplicate by titrations of
1l
M
active papain or S-(methylthio)papain with the
variants. The binding to active papain was monitored
by following the decrease in activity of the enzyme with
a chromogenic substrate [38], whereas the binding to
S-(methylthio)papain was monitored by following the
change in tryptophan fluorescence accompanying the
interaction [41]. The binding stoichiometries were deter-
mined by nonlinear least-squares regression analysis of the
titration curves [41].
Inhibition constants
Apparent inhibition constants, K
i
,
app
, for the inhibition of
cathepsins L and B by the cystatin A mutants were obtained
from the equilibrium rates of hydrolysis of a fluorogenic
substrate by the enzyme at different inhibitor concentrations
Table 1. Primers for construction of expression vectors for cystatin A second-loop mutants. All sequences are given in the 5¢fi3¢direction. Codons
for Gly, replacing residues to be mutated, are underlined, and base changes introducing the mutations are in bold.
Primer Mutation Direction Sequence
Standard All Forward GCTCAGGCGACCATGGGCCATCATCATC
Reverse CTTGCATGCCCTGCAGGTCG
Mutagenic L73G Forward GTATTCAAAAGTGGTCCCGGACAAAATGAGGACTTG
Reverse TCCGGGACCACTTTTGAATACTTTCAAGTGCATATATTTATT
P74G Forward CAAAAGTCTTGGCGGACAAAATGAGGACTTGGTAC
Reverse CATTTTGTCCGCCAAGACTTTTGAATACTTTCAAGTGC
Q76G Forward CTTCCCGGAGGAAATGAGGACTTGGTACTTACTG
Reverse CCTCATTTCCTCCGGGAAGACTTTTGAATAC
N77G Forward CGGACAAGGTGAGGACTTGGTACTTACTGGATAC
Reverse CAAGTCCTCACCTTGTCCGGGAAGACTTTTG
FEBS 2002 Second protease-binding loop of cystatin A (Eur. J. Biochem. 269) 5651

[28,32]. Product formation was continuously monitored in a
conventional fluorimeter (F-4000; Hitachi, Tokyo, Japan)
as in previous work [28]. The substrates were carbobenz-
oxy-
L
-phenylalanyl-
L
-arginine 4-methylcoumaryl-7-amide
(Peptide Institute, Osaka, Japan) for cathepsin L and
carbobenzoxy-
L
-arginyl-
L
-arginine 4-methylcoumaryl-
7-amide (Peptide Institute) for cathepsin B at concentra-
tions of 5 and 10 l
M
, respectively. The fluorescence never
exceeded that corresponding to 5% substrate hydrolysis.
Inhibitor concentrations were at least 10-fold higher than
enzyme concentrations. The inhibition of the enzymes by
L73G-cystatin A was analysed at cystatin concentrations
ranging from (0.1–0.5) ·K
i,app
to (6–10) ·K
i,app
. Corres-
ponding measurements with P74G-cystatin A were per-
formed at inhibitor concentrations varying from
(0.5–2) ·K
i,app
to (10–14) ·K
i,app
, whereas the range was
from (3–4) ·K
i,app
to (10–30) ·K
i,app
for Q76G-cystatin A
and N77G-cystatin A. Values of K
i,app
were derived by
nonlinear regression analyses of plots of the ratio between
the inhibited and uninhibited rates of substrate hydrolysis
against inhibitor concentration [32]. True inhibition con-
stants, K
i
, were obtained after correction for substrate
competition [32,45,46].
Association kinetics
Association rate constants, k
ass,
for the inhibition of papain
and cathepsins L and B by the cystatin A mutants were
determined by continuously monitoring the loss of enzyme
activity in the presence of a fluorogenic substrate in either a
conventional fluorimeter (see above) or a stopped-flow
fluorimeter (SX-17
MV
; Applied Biophysics, Leatherhead,
UK) [28,38]. The substrate for papain was 10 l
M
carbo-
benzoxy-
L
-phenylalanyl-
L
-arginine 4-methylcoumaryl-7-
amide (Peptide Institute), and the substrates for cathepsins
L and B and their concentrations were the same as those
used to determine K
i
(see above). The fluorescence was
always lower than that given by 5% substrate hydrolysis.
The concentrations of the inhibitors were at least 10-fold
higher than those of the enzymes and were varied in a 10–
20-fold range. The highest inhibitor concentrations in
reactions with papain and cathepsin L were 10–20 n
M
,
whereas reactions with cathepsin B were analyzed at
inhibitor concentrations up to 30 l
M
for L73G-cystatin A
andupto0.3–0.5l
M
for the other mutants. Apparent
pseudo-first-order rate constants, k
obs,app
, were obtained by
nonlinear least-squares regression analysis of the progress
curves [28]. Apparent association rate constants, k
ass,app
,
were calculated from the slopes of plots of k
obs,app
vs.
inhibitor concentration and were corrected for substrate
competition to give the true association rate constants, k
ass
[28,45–47].
Dissociation kinetics
Dissociation rate constants, k
diss
, for the complexes of the
cystatin A mutants with papain were determined by dis-
placement experiments, essentially as detailed previously
[14,16]. Papain dissociating from the complexes was trapped
by a high excess of chicken cystatin (form 2), which binds
faster and more tightly to papain than cystatin A or the
cystatin A mutants do [14,18] (see also Results) and thereby
prevents reassociation of the cystatin A variants with the
enzyme. The concentration of the cystatin A mutant–
papain complexes was 2.5–5.0 l
M
, and the molar ratio of
the displacing chicken cystatin to the complexes varied
between 10-fold and 50-fold. The progress of the reaction
was monitored for 100–150 h by following the appearance
of the newly formed complex between papain and chicken
cystatin, analyzed by ion-exchange chromatography on
a MonoQTM column (Amersham Biosciences, Uppsala,
Sweden). Form 2 of chicken cystatin was used because its
lower isoelectric point allows the complex with papain to be
well separated and thus easily quantified in this analysis.
k
diss
was calculated as described previously [14].
k
diss
for the complex between L73G-cystatin A and
cathepsin L was determined by trapping the enzyme
dissociated from the complex by a high concentration of
the substrate, carbobenzoxy-
L
-phenylalanyl-
L
-arginine
4-methylcoumaryl-7-amide, which binds tightly to cathep-
sin L with a K
m
of 1.8 l
M
[45]. In most experiments, the
complex was formed by incubating 0.04 n
M
cathepsin L
with 0.4 n
M
L73G-cystatin A for 90 min, which resulted in
an essentially complete reaction, with 80% of the enzyme
being saturated with the inhibitor. The substrate was then
added to a final concentration of 100 l
M
with minimal
dilution of the complex. Alternatively, the complex was
formed by incubation of 1 n
M
cathepsin L with 10 n
M
L73G-cystatin A for 15 min, resulting in 99% of the
enzyme being bound in the complex, and this mixture was
then diluted 1000-fold into 100 l
M
substrate. In both cases,
the dissociation of the complex was monitored in a
conventional fluorimeter by continuously recording the
fluorescence increase due to cleavage of the substrate by the
liberated cathepsin L. The fluorescence never exceeded that
corresponding to 5% substrate hydrolysis. k
diss
was deter-
mined by nonlinear least-squares regression analysis of the
progress curves [15].
Fluorescence emission spectroscopy
Fluorescence emission spectra of free papain and wild-type
or L73G-cystatin A, as well as of complexes of papain with
either of the two cystatin A variants, were recorded in an
SLM 4800S spectrofluorimeter (SLM-Aminco, Urbana, IL,
USA) with an excitation wavelength of 280 nm, as
described previously [16,41]. Papain and cystatin concen-
trations were 1.0 and 1.2 l
M
, respectively, giving > 99%
saturation of enzyme with inhibitor in analyses of the
complexes. All spectra were corrected for inner-filter effects
and for the wavelength dependence of the instrumental
response [41] and were normalized to a fluorescence
intensity of 1.0 for free papain at the wavelength of the
emission maximum. Difference spectra between the com-
plexes and the free proteins were calculated as in [41].
Protein modeling
The structure of human cystatin A in complex with active
papain was modeled on to the X-ray structure of the
complex between human C3S-cystatin B and S-(carboxy-
methyl)papain (PDB entry 1STF) [13] with the program
SWISS
-
PDB
Viewer (http://www.expasy.ch/spdbv/). The most
favorable rotamers of the side chains of the 46 residues
of cystatin A which differ from those of cystatin B [1]
were initially selected by the program [48], and the
5652 A. Pavlova et al.(Eur. J. Biochem. 269)FEBS 2002

S-carboxymethyl group of the papain moiety of the
complex was removed in the same manner. The model
was then corrected by the program facility Quick and Dirty
Fixingof all side chains in the complex, followed by
Exhaustive Search Fixingof the side chains within the
Leu73–Asn77 segment in the second binding loop of
cystatin A. After each of these steps, the conformation of
the second binding loop in the complex was refined by
energy minimization of the Leu73–Asn77 segment and all
neighboring residues within 6 A
˚. The possibility of other
residues replacing Gly75 in the final model was evaluated by
Quick and Dirty Fixingof all side chains in the complex
after each replacement.
Miscellaneous procedures
For N-terminal sequencing and determination of molecular
masses, the mutants were first desalted into 0.1% (v/v)
trifluoroacetic acid by gel chromatography on Fast-Desalting
PC 3.2/10 columns (Amersham Biosciences). N-Terminal
sequences were analyzed by Edman degradation in an
Applied Biosystems 477A Protein Sequencer. Molecular
masses were measured by MALDI MS in a Kratos Kompact
MALDI 4 instrument (Kratos, Manchester, UK) as in
[18]. SDS/PAGE under reducing and nonreducing condi-
tions was performed with the Tricine buffer system [49].
Experimental conditions
All equilibrium and kinetic experiments were performed at
25.0 ± 0.2 C. The proteases were first activated by 1 m
M
dithiothreitol in the reaction buffer for 10 min at 25 C. The
inhibition of papain was studied in 50 m
M
Tris/HCl,
pH 7.4, containing 100 m
M
NaCl, 0.1 m
M
EDTA and,
except in the displacement experiments, 1 m
M
dithiothreitol
and 0.01% (w/v) Brij35. The interaction with cathepsin L
wasanalyzedin100 m
M
sodium acetate, pH 5.5, containing
100 m
M
NaCl, 1 m
M
EDTA, 1 m
M
dithiothreitol, and
0.01% (w/v) Brij35, whereas the buffer for cathepsin B
was 50 m
M
Mes/NaOH, pH 6.0, containing 100 m
M
NaCl,
0.1 m
M
EDTA, 1 m
M
dithiothreitol, and 0.1% (w/v)
poly(ethylene glycol) 6000.
RESULTS
Preparation, homogeneity and activity of cystatin A
mutants
Four variants of cystatin A, each with a single amino-acid
residue, Leu73, Pro74, Gln76 or Asn77, substituted by Gly
were produced by recombinant DNA techniques. All these
mutations are in the most exposed part of the second
protease-binding loop of the inhibitor (Fig. 1A). Residue 75
was not substituted, as it is Gly in the wild-type sequence.
The expression vectors were constructed by PCR-based site-
directed mutagenesis and contained the expected mutant
sequences in the case of the L73G, P74G and N77G
mutants. However, all vectors for the Q76G mutant purified
from 18 individual clones had, in addition to the desired
mutation, a T fiC substitution in the codon for Thr83.
This substitution was in the region specified by the forward
mutagenic primer for this mutant and was probably due to
an erroneously synthesized primer. As this additional
mutation is silent, one of the isolated vectors was neverthe-
less used for expression of Q76G-cystatin A. The mutants
were expressed with a removable His-tag and with a signal
peptide directing the proteins to the periplasmic space of
E. coli, facilitating purification.
All purified mutants were > 99.5% homogeneous on
SDS/PAGE. N-Terminal sequencing of the first five
residues confirmed that the His-tag was cleaved off properly
by enterokinase for all mutants. The molecular masses,
determined by MS, corresponded within 4.5 Da to those
calculated from the expected amino-acid sequences, con-
firming the correct length of the mutants, as well as the
presence of the desired mutations. All mutants bound active
papain and S-(methylthio)papain with stoichiometries
between 0.95 and 1.0, i.e. they were essentially fully active
in inhibition of cysteine proteases.
Binding affinity
All four cystatin A mutants bound so tightly to papain that
the affinity of the binding could not be determined by
equilibrium methods, because of the instability of the
enzyme at the low concentrations and the long reaction
times that would have been necessary. Therefore, K
d
for the
interaction with papain was calculated as k
diss
/k
ass
from
independently measured rate constants (see below and
Table 2), as was K
d
for wild-type cystatin A binding to this
enzyme in previous work [18]. Only the L73G mutation
caused a pronounced, 300-fold, decrease in the affinity for
papain, compared with that of the wild-type inhibitor
(Table 2). In contrast, the P74G, Q76G, and N77G muta-
tions resulted in minimal, less than twofold, changes in
affinity.
The high affinity of most mutants for cathepsin L also
precluded an accurate determination of K
i
from equilibrium
measurements. Such experiments gave only upper limits of
K
i
for the interaction of P74G, Q76G, and N77G cystatin A
with this protease (Table 2), similar to previous analyses of
K
i
for the wild-type inhibitor [35]. Moreover, as k
diss
for
these tight interactions could not be determined (see below),
K
d
could not be calculated from the rate constants. No
meaningful comparisons of the affinities of the three
mutants for cathepsin L with that of wild-type cystatin A
were therefore possible. However, a reliable K
i
for the
inhibition of cathepsin L by L73G-cystatin A was obtained
by equilibrium measurements and was > 10-fold higher
than that for wild-type cystatin A. The measured K
i
for this
mutant agreed well with K
d
calculated from k
ass
and k
diss
(see below and Table 2).
K
i
for the inhibition of cathepsin B by all cystatin A
forms was sufficiently high to be well determined by
equilibrium analyses. The L73G mutation caused a sub-
stantial, 4000-fold, increase in K
i
which was confirmed by
calculations of K
d
from k
ass
and k
diss
(Table 2). A smaller,
10-fold, increase in K
i
for cathepsin B was also observed
for the P74G mutant, whereas the affinities of both Q76G
and N77G cystatin A for the enzyme differed minimally,
about twofold, from that of wild-type cystatin A (Table 2).
Association rate constants
The kinetics of association of the cystatin A mutants
with papain, cathepsin L and cathepsin B were analyzed
FEBS 2002 Second protease-binding loop of cystatin A (Eur. J. Biochem. 269) 5653


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)