
Phospholipase C, protein kinase C, Ca
2+
/calmodulin-dependent
protein kinase II, and redox state are involved in epigallocatechin
gallate-induced phospholipase D activation in human astroglioma cells
Shi Yeon Kim
1
, Bong-Hyun Ahn
1
, Joonmo Kim
1
, Yoe-Sik Bae
2
, Jong-Young Kwak
2
, Gyesik Min
3
,
Taeg Kyu Kwon
4
, Jong-Soo Chang
5
, Young Han Lee
6
, Shin-Hee Yoon
1
and Do Sik Min
1
1
Department of Physiology, College of Medicine, The Catholic University of Korea, Seoul, Korea;
2
Medical Research Center for
Cancer Molecular Therapy and Department of Biochemistry, College of Medicine, Dong-A University, Busan, Korea;
3
Department of
Microbiological Engineering, Jinju National University, Korea;
4
Department of Immunology, School of Medicine, Keimyung
University, Daegu, Korea;
5
Department of Life Science, Daejin University, Kyeongggido, Korea;
6
Division of Molecular and Life
Science, College of Science and Technology, Hanyang University, Ansan, Korea
We show that epigallocatechin-3 gallate (EGCG), a major
component of green tea, stimulates phospholipase D (PLD)
activity in U87 human astroglioma cells. EGCG-induced
PLD activation was abolished by the phospholipase C
(PLC) inhibitor and a lipase inactive PLC-c1mutant,which
is dependent on intracellular or extracellular Ca
2+
,withthe
possible involvement of Ca
2+
/calmodulin-dependent pro-
tein kinase II (CaM kinase II). EGCG induced translocation
of PLC-c1 from the cytosol to the membrane and PLC-c1
interaction with PLD1. EGCG regulates the activity of PLD
by modulating the redox state of the cells, and antioxi-
dants reverse this effect. Moreover, EGCG-induced PLD
activation was reduced by PKC inhibitors or down-regula-
tion of PKC. Taken together, these results show that, in
human astroglioma cells, EGCG regulates PLD activity
via a signaling pathway involving changes in the redox state
that stimulates a PLC-c1 [Ins(1,4,5)P
3
-Ca
2+
]–CaM kinase
II–PLD pathway and a PLC-c1 (diacylglycerol)–PKC–PLD
pathway.
Keywords:Ca
2+
/calmodulin-dependent protein kinase II;
epigallocatechin-3 gallate; phospholipase C-c1; phospho-
lipase D; reactive oxygen species.
Phospholipase D (PLD) catalyzes the hydrolysis of the most
abundant membrane phospholipid, phosphatidylcholine, to
generate phosphatidic acid and choline and is assumed to
have an important function in cell regulation [1]. Signal-
dependent activation of PLD has been demonstrated in
numerous cell types stimulated by various hormones,
growth factors, cytokines, neurotransmitters, adhesion
molecules, drugs, and physical stimuli [2]. Pathways leading
to PLD activation include protein serine/threonine kinases,
e.g. protein kinase C (PKC), small GTPases, e.g. ADP-
ribosylation factor, RhoA and Ral, phosphatidylinositol
4,5-bisphosphate, and tyrosine kinases [2–4]. To date, two
distinct isoforms of mammalian PLD have been cloned,
PLD1 and PLD2. These isoforms share about 50% amino
acid similarity, but exhibit quite different regulatory prop-
erties [5,6]. Both proteins appear to be complexly regulated,
usually in an agonist-specific and cell-specific manner, and
the molecular mechanisms underlying their functions have
not been fully elucidated.
Green tea (Camellia sinensis) is a popular beverage world
wide, and its possible health benefits have received a great
deal of attention. Documented beneficial effects of green tea
and its active components include cancer chemoprevention,
inhibition of the growth, invasion and metastasis of tumor
cells, as well as antiviral and anti-inflammatory activities [7].
Green tea contains the characteristic polyphenolic com-
pounds epigallocatechin-3-gallate (EGCG), epigallocate-
chin (EGC), epicatechin-3-gallate (ECG) and epicatechin
(EC). EGCG is considered to be the constituent primarily
responsible for the green tea effects [8,9]. Although the
activity of EGCG in some biological events has been
investigated, its effect on the signal transduction cascade is
not yet fully defined. Recently, it has been reported that
EGCG produces reactive oxygen species (ROS) including
H
2
O
2
[10]. Oxidant-induced PLD activation and redox
regulation of PLD have been reported in a variety of cells
such as Swiss 3T3 fibroblasts [11], PC12 cells [12,13], and
endothelial cells [14]. ROS such as H
2
O
2
and superoxide
have been shown to be generated in a variety of cells
stimulated with cytokines, growth factors, and agonists of
Correspondence to D. S. Min, Department of Molecular Biology,
College of Natural Science, Pusan National University,
Geumjeong-gu, Busan 609-735, Korea. Fax: +82 51 513 9258,
Tel.: +82 51 510 1775 (from 1 September 2004).
Abbreviations: CaM kinase II, Ca
2+
/calmodulin-dependent protein
kinase II; DCFH, 2¢,7¢-dichlorofluorescein diacetate; DCF, 2¢,7¢-
dichlorofluorescein; DMEM, Dulbecco’s modified Eagle’s medium;
EC, epicatechin; ECG, epicatechin-3-gallate; EGC, epigallocatechin;
EGCG, epigallocatechin-3-gallate; PKC, protein kinase C:
PLC, phospholipase C; PLD, phospholipase D; PtdBut,
phosphatidylbutanol; ROS, reactive oxygen species.
(Received 29 March 2004, revised 25 May 2004,
accepted 3 June 2004)
Eur. J. Biochem. 271, 3470–3480 (2004) FEBS 2004 doi:10.1111/j.1432-1033.2004.04242.x

G protein-linked receptors, and it has been suggested that
they may act as second messengers [15]. However, no
information is available on how EGCG affects PLD-
mediated signaling pathways. Therefore, we investigated
PLD regulation by EGCG.
We show that EGCG significantly stimulates PLD
activity and that EGCG-induced PLD activation is medi-
ated via a signaling pathway involving redox-dependent
changes in the cell, which stimulate the PLC-c1
[Ins(1,4,5)P
3
–Ca
2+
]–Ca
2+
/calmodulin-dependent protein
kinase II (CaM kinase II)–PLD pathway and the PLC-c1
(diacylglycerol)–PKC–PLD pathway.
Experimental procedures
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine
serum and LipofectAMINE were purchased from Invitro-
gen. EGCG, EGC, ECG and EC were obtained from Sigma.
Protein A–Sepharose was from Amersham Biosciences
Biotech. Antibody to PLC-c1 was from Upstate Biotechno-
logy. PD98059, U-73122, U-73343, Ro-31-8220, and
calphostin C were purchased from Biomol Research
Laboratories (Plymouth Meeting, PA, USA). KN-92,
KN-93, sphingosine 1-phosphate, and pertussis toxin were
obtained from Calbiochem. Other chemicals were purchased
from Sigma. Rabbit polyclonal antibody that recognizes
both PLD1 and PLD2 was generated as described previously
[16]. Authentic phosphatidylbutanol (PtdBut) standard
was from Avanti Polar Lipid. myo-[2-
3
H]Inositol and
[9,10-
3
H]myristate were purchased from Perkin-Elmer Life
Sciences. AG 1-X8 anion-exchange resin was bought from
Bio-Rad. Silica gel 60A TLC plates were from Whatman.
Horseradish peroxidase-conjugated anti-mouse IgG and
anti-rabbit IgG were from Kirkegaard and Perry Laboratory
(Gaithersburg, MD, USA). The ECL Western blotting
detection kit was from Amersham Biosciences Biotech.
Cell culture and transfection
U87 human astroglioma were maintained in DMEM
supplemented with 10% (v/v) fetal bovine serum under
5% CO
2
. Cells were transiently transfected for 40 h with
plasmids encoding empty vector, PLD1, PLD2, or a lipase
inactive mutant PLC-c1 (H335Q) expression vectors using
LipofectAMINE according to the manufacturer’s instruc-
tions.
Measurement of phosphoinositide hydrolysis by PLC
The cells were labeled with myo-[2-
3
H]inositol (2 lCiÆmL
)1
)
in inositol-free DMEM for 20 h. Subsequently, the labeled
cells were pretreated with 20 m
M
LiCl for 15 min. After
stimulation with EGCG, the reaction was terminated by the
addition of ice-cold 5% HClO
4
. The extracts were applied
toaBio-RadDowexAG1-X8anion-exchangecolumn.
The column was then washed with 10 mL distilled water
followedby10mL60m
M
ammonium formate containing
5m
M
sodium tetraborate. Total inositol phosphates were
eluted with a solution containing 1
M
ammonium formate
and 0.1
M
formic acid.
PLD assay
PLD activity was assessed by measuring the formation of
[
3
H]PtdBut, the product of PLD-mediated transphosphati-
dylation, in the presence of butan-1-ol. Cells were sub-
cultured in six-well plates at 2 ·10
5
cells per well and
serum-starved in the presence of 1 lCiÆmL
)1
[
3
H]myristic
acid. After overnight starvation, the cells were washed three
times with 5 mL NaCl/P
i
and pre-equilibrated in serum-free
DMEM for 1 h. For the final 10 min of preincubation,
0.3% butan-1-ol was included. At the end of the preincu-
bation, cells were treated with agonists for the indicated
times. The extraction and characterization of lipids by TLC
were performed as described previously [16].
Subcellular fractionation
Serum-starved cells were treated with 500 l
M
EGCG for
10 min, and washed with NaCl/P
i
and harvested by
microcentrifugation. The cells were then resuspended in
lysis buffer (20 m
M
Hepes, pH 7.4, 10% glycerol, 1 m
M
EDTA, 1 m
M
EGTA, 1 m
M
dithiothreitol, 1 m
M
phenyl-
methanesulfonyl fluoride and 10 lgÆmL
)1
leupeptin) and
lysed by 20 passages through a 25-gauge needle. Trypan
blue staining of the lysate indicated > 95% disruption of
the cells. The lysates were then spun at 100 000 gfor 1 h at
4C to separate the cytosolic and membrane fractions.
Membrane fractions were washed twice with the buffer to
remove cytosolic proteins.
Digital calcium imaging
Intracellular calcium was measured as described previously
[17]. Cells were plated on to glass coverslips and loaded with
2l
M
fura-2 acetoxymetyl ester (Molecular Probes) for
45 min at 37C. The coverglass was then mounted in a flow-
through chamber. The chamber containing the fura-2-
labeled cells was mounted and alternately excited at 340 or
380 nm. Digital fluorescence images were collected with a
cooled CCD camera. [Ca
2+
]
i
was calculated from the ratio
of the two background-subtracted digital images. Ratios
were converted into free [Ca
2+
]
i
by the equation
½Ca2þi¼KbðRRmin Þ=ðRmax RÞ
in which Ris the 340/380-nm fluorescence emission ratio
and K¼224 n
M
, the dissociation constant for fura-2 [18].
Immunoprecipitation
U87 cells were harvested and lysed with lysis buffer (20 m
M
Hepes, pH 7.2, 1% Triton X-100, 1% sodium deoxycho-
late, 0.2% SDS, 150 m
M
NaCl, 1 m
M
Na
3
VO
4
,1m
M
NaF,
10% glycerol, 10 lgÆmL
)1
leupeptin, 10 lgÆmL
)1
aprotinin,
1m
M
phenymethanesulfonyl fluoride). The cells were then
centrifuged at 10 000 gfor 1 h, and the resulting superna-
tant was incubated with antibody to PLD or PLC-c1and
Protein A–Sepharose for 4 h at 4 C with rocking. Protein
concentrations were determined using the Bio-Rad Protein
Assay with BSA as standard. The immune complexes were
collected by centrifugation and washed five times with
buffer (20 m
M
Tris/HCl, pH 7.5, 1 m
M
EDTA, 1 m
M
FEBS 2004 Regulation of phospholipase D by EGCG (Eur. J. Biochem. 271) 3471

EGTA, 150 m
M
NaCl, 2 m
M
Na
3
VO
4
, 10% glycerol and
1% Nonidet P40) and resuspended in sample buffer. The
final pellet was loaded on to a polyacrylamide gel for
immunoblot analysis.
Immunoblot analysis
Proteins were denatured by boiling for 5 min at 95 Cin
Laemmli sample buffer [19], separated by SDS/PAGE,
and transferred to nitrocellulose membranes. After being
blocked in Tris/Tween-buffered saline containing 5%
skimmed milk powder, the membranes were incubated with
individual monoclonal or polyclonal antibodies and then
further incubated with anti-mouse or anti-rabbit IgG
coupled to horseradish peroxidase. Blots were detected
using the enhanced chemiluminescence kit according to the
manufacturer’s instructions.
Confocal immunofluorescence microscopy
U87 cells grown on poly(
L
-lysine)-coated glass coverslips
were serum-starved for 24 h. After stimulation with EGCG,
the cells were fixed in 3.7% (w/v) formaldehyde for 15 min
and quenched using 50 m
M
NH
4
Cl for 10 min. After
permeabilization using 1% Triton X-100 for 5 min, the cells
were incubated with blocking buffer (1% goat serum in
NaCl/P
i
) at room temperature for 1 h, and then with
primary antibody overnight at 4 C,andthenwith
subclass-specific secondary antibodies [fluorescein isothio-
cyanate-conjugated donkey anti-(mouse IgG) (Jackson
ImmunoResearch, West Grove, PA, USA) or Texas
Red-conjugated goat anti-(rabbit IgG) (Jackson
ImmunoResearch)] for 1 h. After being washed, the cover-
slips were mounted on to slides in Prolong (Molecular
Probes). Images in the Figures were acquired using a Zeiss
MRC 1024 microscope (Bio-Rad).
Detection of intracellular ROS generation
Intracellular ROS production was monitored using 2¢,7¢-
dichlorofluorescein diacetate (DCFH) (Sigma-Aldrich),
which is oxidized to the fluorescent product 2¢7¢-dichloro-
fluorescein (DCF) by ROS [20]. Briefly, U87 cells grown on
coverslips were loaded with ROS-sensitive dye (10 l
M
).
After 15 min at room temperature, the cells were washed
three times with serum-free medium, and treated with
vehicle alone or EGCG. ROS produced were monitored
using an excitation wavelength of 490 nm and emission
fluorescence at 520 nm with a confocal Microscope (Zeiss).
Determination of glutathione concentration
Cells treated with EGCG were washed in NaCl/P
i
and then
scraped into 5% metaphosphoric acid. Reduced glutathione
(GSH) was quantified using a commercially available GSH
determination kit (Calbiochem). Briefly, the method was
basedonachemicalreactionwhichproceededintwosteps.
The first step led to the formation of substitution products
(thioethers) between 4-chloro-1-methyl-7-trifluromethyl-
quinolinum methylsulfate and all mercaptans which were
present in the sample. The second step included a
b-elimination reaction under alkaline conditions. This
reaction was mediated by 30% NaOH which specifically
transformed the substituted product (thioether) obtained
with GSH into a chromophoric thione.
Results
EGCG stimulates PLD activity in U87 human
astroglioma cells
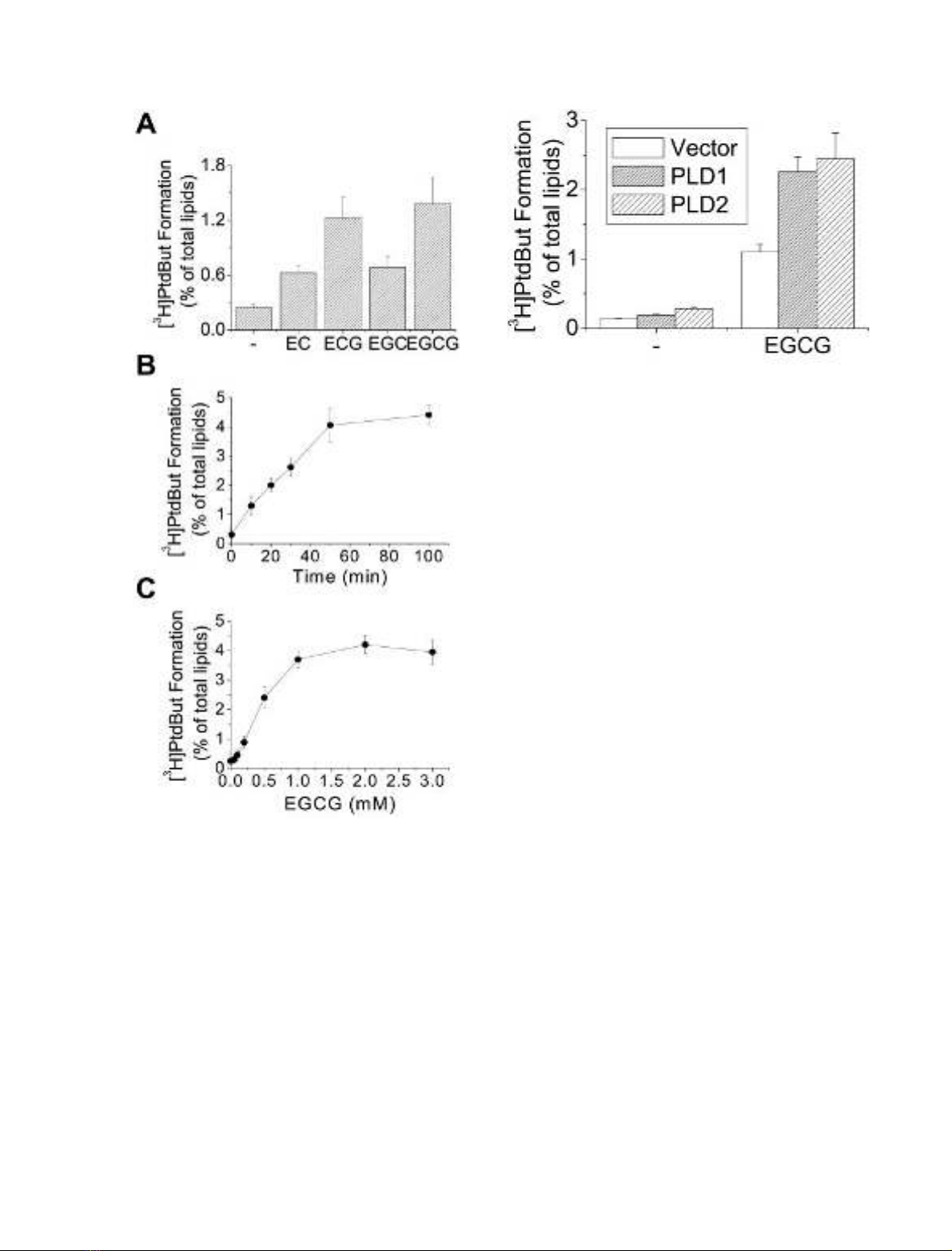
We investigated whether green tea polyphenols activate
PLD in U87 human astroglioma cells. Cells were treated
for 30 min with EGCG, ECG, EGC or EC. The data
presented in Fig. 1A show that these polyphenolic com-
pounds significantly stimulated PLD activity, with EGCG
being the most potent activator. EGCG-induced [
3
H]Ptd-
But formation increased in a time- and concentration-
dependent manner (Fig. 1B,C). Activation of PLD by
EGCG continued up to 50 min and then remained
constant up to 100 min; maximum activation was
observed at 1 m
M
EGCG. Using PLD antibodies, we
detected PLD1, but not PLD2, in U87 cells. However,
transient transfection of cells with PLD1 and PLD2
expression vectors revealed that EGCG activates both
PLD1 and PLD2 (Fig. 2).
Role of PLC in EGCG-induced PLD activation
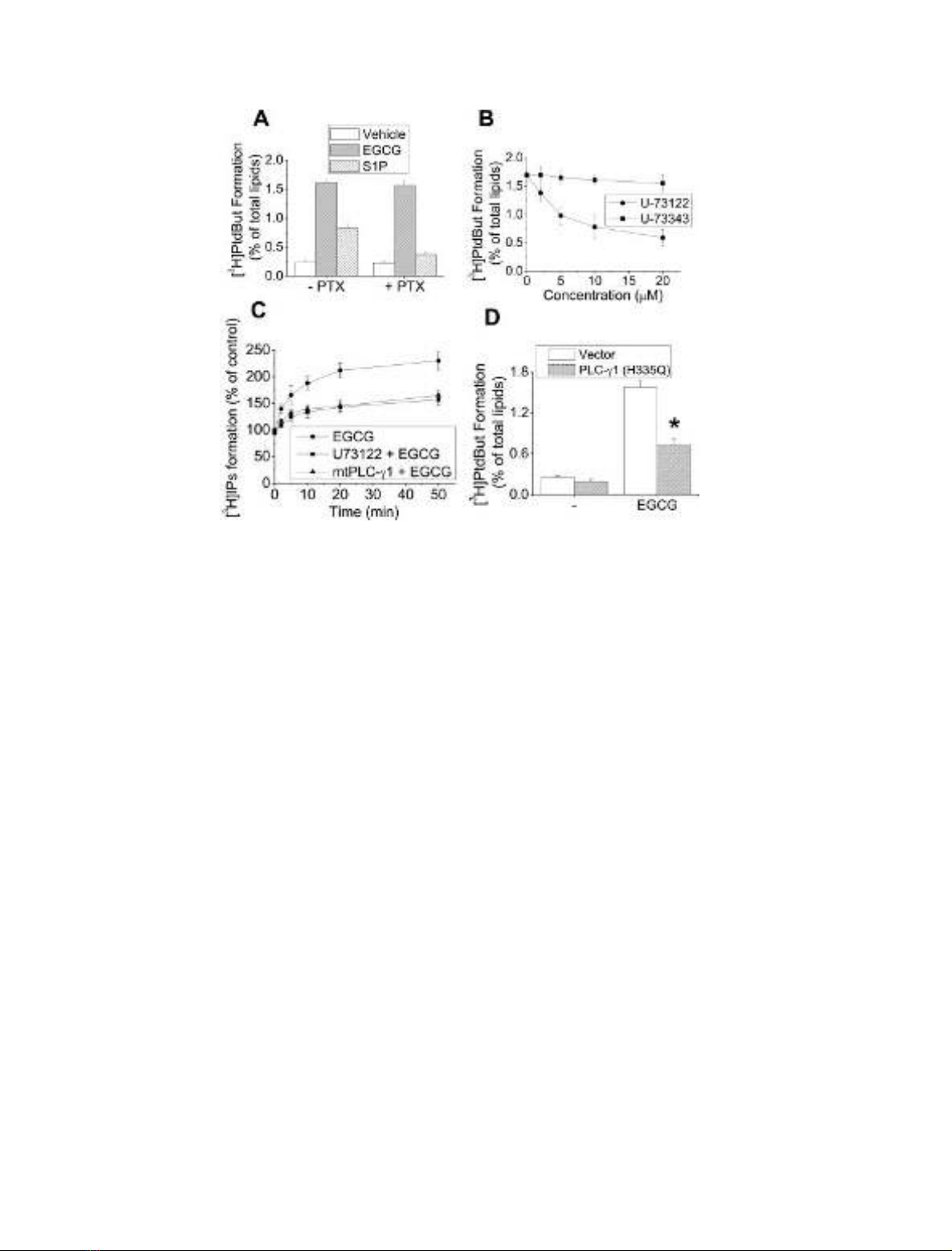
Numerous studies have implicated PLC in the activation of
PLD [21,22]; however, the results of other studies have
suggested that PLC is not involved [23,24]. To determine
whether PLC activity or G-protein-mediated signaling was
involved in EGCG-induced PLD activation in U87 cells, we
examined the effects of pertussis toxin and the phospho-
inositide-specific PLC inhibitor, U-73122. Pretreatment
with pertussis toxin (100 ngÆmL
)1
for 24 h) inhibited
sphingosine 1-phosphate-induced PLD activation, suggest-
ing that this activation reaction is dependent on the
G
i
protein-mediated signaling response in these cells. How-
ever, pertussis toxin had no effect on EGCG-induced PLD
activation (Fig. 3A). EGCG-induced PLD activation was
significantly attenuated by the PLC-specific inhibitor U-
73122, in a dose-dependent manner, but not by its inactive
analog U-73343 (Fig. 3B). These data suggest that phos-
phoinositide-specific PLC activation via a pertussis toxin-
insensitive pathway plays a critical role in EGCG-induced
PLD activity in these cells. We also investigated whether
EGCG induces PLC activity in U87 cells. The data
presented in Fig. 3C show that EGCG treatment stimulates
PLC activity, as measured by formation of [
3
H]inositol
phosphates, which peaked after 10 min and was sustained
for at least 50 min. In a control experiment, the PLC
inhibitor U73122 actually inhibited PLC activity in cells
stimulated by EGCG (Fig. 3C). We found that PLC-c1was
the predominantly expressed PLC in U87 cells, indicating
that the PLC activity shown in these cells may be due mainly
to PLC-c1. We found that ectopic expression of the lipase
inactive mutant PLC-c1(His335fiGln) [25] attenuated
endogenous PLC activity by EGCG, suggesting surprising
effectiveness of the catalytically inactive PLC-c1mutant
expression plasmid on the suppression of EGCG-stimulated
PLC activity. Therefore, we examined the involvement of
PLC-c1 in the PLD activation by EGCG in U87 cells.
3472 S. Y. Kim et al.(Eur. J. Biochem. 271)FEBS 2004
Interestingly, expression of the lipase inactive mutant PLC-
c1 significantly attenuated EGCG-induced PLD activation
(Fig. 3D), suggesting that PLC-c1 is involved in this
process.
EGCG induces a rise in [Ca
2+
]
i
in U87 cells
As EGCG stimulates PLC activity, it might induce an
increase in [Ca
2+
]
i
in U87 cells. [Ca
2+
]
i
after EGCG
treatment was visualized by loading the cells with Fura-2/
AM. Figure 4 shows simultaneous measurement of [Ca
2+
]
i
increases in different cells, using digital calcium imaging.
One trace represents [Ca
2+
]
i
increase in one cell, and the
different traces represent each [Ca
2+
]
i
increase pattern in the
different cells. The rise in [Ca
2+
]
i
after EGCG stimulation
peaked within 3 min and then decreased (Fig. 4A). An
EGCG-stimulated increase in [Ca
2+
]
i
may result from an
influx of extracellular calcium. To test this possibility, we
treated cells with EGCG in the presence of Ca
2+
-free
buffer. The level of [Ca
2+
]
i
after EGCG treatment was
visualized by loading the cells with Fura-2/AM. For cells in
Ca
2+
-free buffer, EGCG caused only a very small increase
in [Ca
2+
]
i
(Fig. 4B). These results clearly show that
treatment of U87 cells with EGCG results in an increase
in cytosolic calcium. Furthermore, the results suggest that
an influx of calcium from the extracellular medium is mainly
responsible for this rise.
EGCG induces translocation of PLC-c1 and its interaction
with PLD1
After growth factor stimulation, PLC-c1istranslocated
from the cytosol to the membrane, where its substrate
molecules reside [26]. We examined whether EGCG
induced PLC-c1 translocation. Incubation with EGCG
for 10 min significantly increased the amount of PLC-c1
associated with the membrane fraction in U87 cells
(Fig. 5A). Using confocal immunofluorescence microscopy,
we confirmed that PLC-c1 translocation to membrane
regions increased after EGCG treatment. Furthermore,
colocalization of PLD1 and PLC-c1increasedinthe
membraneous region after EGCG stimulation (Fig. 5B).
We sought to confirm this apparent interaction between
PLD1 and PLC-c1 in EGCG-stimulated U87 cells. We
found that PLD1 showed a mild interaction with PLC-c1in
unstimulated cells, and this association increased after
treatment of EGCG for 10 min (Fig. 5C). These data
suggest that PLD1 associates with PLC-c1 during EGCG-
induced PLD activation.
Fig. 2. EGCG activates both PLD1 and PLD2. U87 cells were tran-
siently transfected for 40 h with plasmids encoding empty vector,
PLD1, or PLD2 expression vectors using LipofectAMINE according
to the manufacturer’s instructions, labeled with [
3
H]myristic acid, and
treated with EGCG (500 l
M
) for 30 min. PLD activity was measured
as described in Experimental procedures. Results are means ± SD
from three independent experiments.
Fig. 1. Green tea polyphenols stimulate PLD activity in U87 human
astroglioma cells. Cells were cultured in six-well plates, labeled with
[
3
H]myristate, and treated for 30 min without or with 500 l
M
EC,
ECG, EGC, or EGCG in the presence of 0.3% butanol (A).
[
3
H]Myristate-labeled cells were treated with 500 l
M
EGCG for the
indicated time (B) or with the indicated concentration of EGCG for
50 min (C). The radioactivity incorporated into PtdBut was measured
as described in Experimental procedures. Results are means ± SD
from three independent experiments.
FEBS 2004 Regulation of phospholipase D by EGCG (Eur. J. Biochem. 271) 3473
Pretreatment with antioxidants abolishes activation
of PLC and PLD induced by EGCG
It has been demonstrated that PLC-c1 is activated in
response to oxidant exposure [27,28]. In addition, oxidative
stress stimulates PLD activity in a various cells [11–14].
Therefore, we examined the effect of antioxidants on the
PLC and PLD activation induced by EGCG. Pretreatment
with N-acetylcysteine, a glutathione precursor and scaven-
ger of ROS, decreased EGCG-induced PLC activation in a
dose-dependent manner (Fig. 6A). Moreover, pretreatment
with the antioxidants, catalase and N-acetylcysteine, abol-
ished EGCG-induced PLD activation in a dose-dependent
manner (Fig. 6B,C). These results suggest that EGCG may
increase ROS production and induce activation of PLC and
PLD. Furthermore, we found that incubation of the
astrocytoma cells with H
2
O
2
led to PLD activation
(Fig. 6D). These results demonstrate the role of ROS such
as H
2
O
2
in the EGCG effect on the activation of PLC and
PLD.
EGCG has pro-oxidant activity in U87 astrocytoma cells
It is possible that pro-oxidative activity of EGCG in
astrocytoma cells could explain the activation of PLD. U87
cells were incubated with DCFH to test whether EGCG
increases ROS production. ROS produced in cells causes
oxidation of DCFH, yielding the fluorescent product DCF
[20]. The cells were treated in the presence or absence of
EGCG, and DCF fluorescence was measured (Fig. 7).
EGCG significantly increased fluorescence. This suggests
that EGCG has pro-oxidant activity in astrocytoma cells.
The EGCG-mediated increase in DCF fluorescence was
abolished by pretreating the cells with N-acetylcysteine, a
glutathione precursor and scavenger of ROS (Fig. 7). These
results suggest that EGCG increases ROS production in
U87 cells. We next measured the glutathione (GSH) content
in the cells treated with EGCG in the presence or absence of
N-acetylcysteine to support the redox state of the cells.
EGCG treatment decreased the GSH concentration, and
the decrease in GSH content by EGCG in cells pretreated
with N-acetylcysteine was recovered, suggesting that treat-
ment of cells with EGCG decreases GSH.
EGCG-induced PLD activation is dependent on
intracellular or extracellular Ca
2+
and mediated
by CaM kinase II
Several examples of the participation of Ca
2+
in the
regulation of PLD activity have been reported, although the
effector molecules involved have not been fully character-
ized [29,30]. We found that 1,2-bis-(2-aminophen-
oxy)ethane-N,N,N¢,N¢,-tetra-acetic acid acetoxymethyl
ester (BAPTA/AM), an intracellular chelator of Ca
2+
,
Fig. 3. PLC is involved in EGCG-induced PLD activation. (A) Quiescent U87 cells were pretreated with 200 ngÆmL
)1
pertussis toxin for 24 h,
labeled with [
3
H]myristate,andstimulatedwith1l
M
sphingosine 1-phosphate or 500 l
M
EGCG for 30 min. (B) [
3
H]Myristate-labeled cells were
pretreated with the indicated concentrations of U-73122 or U-73343, and stimulated with EGCG for 30 min. (C) Cells transfected with or without a
catalytically inactive mutant of PLC-c1 (H335Q) were labeled with 1 lCiÆmL
)1
myo-[2-
3
H]inositol, pretreated with or without U-73122 (20 l
M
),
and stimulated with EGCG for the indicated time. PLC activity was measured as described in Experimental procedures. (D) U87 cells were
transiently transfected with a catalytically inactive mutant of PLC-c1 (H335Q), labeled with [
3
H]myristic acid, and treated with EGCG for 30 min.
*P<0.05 compared with cells transfected with vector and treated with EGCG. The radioactivity incorporated into PtdBut was measured as
described in Experimental procedures. Results are means ± SD from three independent experiments.
3474 S. Y. Kim et al.(Eur. J. Biochem. 271)FEBS 2004


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)