
Acylation of lysophosphatidylcholine plays a key role
in the response of monocytes to lipopolysaccharide
Bernhard Schmid
1,2
, Michael J. Finnen
3
, John L. Harwood
1
and Simon K. Jackson
2
1
School of Biosciences, Cardiff University, UK;
2
University of Wales College of Medicine, Cardiff, UK; and
3
Yamanouchi Research Institute, Oxford, UK
Mononuclear phagocytes play a pivotal role in the pro-
gression of septic shock by producing tumor necrosis factor-
a(TNF-a) and other inflammatory mediators in response to
lipopolysaccharide (LPS) from Gram-negative bacteria. Our
previous studies have shown monocyte and macrophage
activation correlate with changes in membrane phospholipid
composition, mediated by acyltransferases. Interferon-c
(IFN-c), which activates and primes these cells for enhanced
inflammatory responses to LPS, was found to selectively
activate lysophosphatidylcholine acyltransferase (LPCAT)
(P< 0.05) but not lysophosphatidic acid acyltransferase
(LPAAT) activity. When used to prime the human mono-
cytic cell line MonoMac 6, the production of TNF-aand
interleukin-6 (IL-6) was approximately five times greater in
cells primed with IFN-cthan unprimed cells. Two LPCAT
inhibitors SK&F 98625 (diethyl 7-(3,4,5-triphenyl-2-oxo2,3-
dihydro-imidazole-1-yl)heptane phosphonate) and YM
50201 (3-hydroxyethyl 5,3¢-thiophenyl pyridine) strongly
inhibited (up to 90%) TNF-aand IL-6 production in
response to LPS in both unprimed MonoMac-6 cells and
in cells primed with IFN-c. In similar experiments, these
inhibitors also substantially decreased the response of both
primed and unprimed peripheral blood mononuclear cells
to LPS. Sequence-based amplification methods showed that
SK&F 98625 inhibited TNF-aproduction by decreasing
TNF-amRNA levels in MonoMac-6 cells. Taken together,
the data from these studies suggest that LPCAT is a key
enzyme in both the pathways of activation (priming) and the
inflammatory response to LPS in monocytes.
Keywords: lysophosphatidylcholine acyltransferase; lyso-
phosphosphatidate acyltransferase; inflammatory response;
lipopolysaccharide.
Endotoxin, the lipopolysaccharide (LPS) component of the
cell walls of Gram-negative bacteria, is an important
microbial molecular pattern that initiates inflammatory
and coagulation reactions as part of the host innate immune
response to infection. However, excessive inflammatory
responses to LPS can lead to septic shock.
Many of the deleterious effects of LPS are mediated by
inflammatory cytokines including tumor necrosis factor
(TNF)-a, interleukin (IL)-1 and IL-6 produced by mono-
nuclear phagocytes (e.g. monocytes and macrophages) [1].
TNF-ahas been shown to be a key mediator in experimen-
tal LPS-induced septic shock as neutralizing antibodies to
TNF-aprevent mortality [2,3] and TNF-areceptor knock-
out mice are less sensitive to the biological effects of LPS
[4]. Understanding and modulating the production of
LPS-induced mediators such as TNF-ahas been the focus
of much research aimed at developing specific therapies for
septic shock.
Studies in our laboratories have been concerned with
elucidating mechanisms of responsiveness to low (nonlethal)
doses of LPS which, we believe, underscore the excessive
inflammatory responses leading to septic shock [5,6]. An
important regulator of LPS-induced biological activity is
interferon (IFN)-c[7,8] and the neutralization of IFN-c
or the deletion of its receptors have been shown to be
protective for the lethal outcomes of several forms of
endotoxic shock [9,10]. An important contribution of IFN-c
to the development of LPS-induced shock is by priming
monocytes/macrophages for increased inflammatory
responsiveness to LPS [11]. The molecular basis for such
priming reactions remain obscure.
In previous studies investigating the priming of mono-
cytes by IFN-c, we showed that this cytokine significantly
modified the phospholipid composition of such cells and
increased the unsaturated fatty acyl groups esterified at the
sn-2 position of phosphatidylcholine (PtdCho) [5,6]. More-
over, we found that IFN-cup-regulated monocyte lysoPC
acyltransferase (LPCAT), a key enzyme in the regulation of
PtdCho fatty acyl composition [6,12]. These results suggest
that LPCAT may regulate the priming of monocytes by
IFN-cand allow increased inflammatory cytokine produc-
tion when these cells are stimulated by LPS. In studies of
T-lymphocyte activation, Szamel and colleagues [13,14]
have demonstrated that activation of the T-cell antigen
Correspondence to S. K. Jackson, Department of Medical Microbio-
logy, University of Wales College of Medicine, Cardiff CF14 4XN,
UK. Fax: + 44 29 20742161, Tel.: + 44 29 20744725,
E-mail: jacksonsk@cardiff.ac.uk
Abbreviations: BSA, bovine serum albumin; DAG, diacylglycerol;
IFN-c, interferon-c; IL-6, interleukin-6; LPS, lipopolysaccharide;
LPAAT, lysophosphatidate acyltransferase; LPCAT, lysophospha-
tidylcholine acyltransferase; NASBATM, nucleic acid sequence based
amplification; PtdOH, phosphatidic acid; PKC, protein kinase C;
PtdCho, phosphatidylcholine; TNF-a, tumor necrosis factor-a.
(Received 11 September 2002, revised 17 March 2003,
accepted 02 May 2003)
Eur. J. Biochem. 270, 2782–2788 (2003) FEBS 2003 doi:10.1046/j.1432-1033.2003.03649.x
receptor/CD3 complex leads to increased incorporation of
polyunsaturated fatty acids into phosphatidylcholine, also
mediated by a LPCAT. Thus, LPCAT may alter the cell
membrane lipid environment so as to favor the assembly of
a signaling complex which can then activate the cellular
response [15].
To elucidate the role of LPCAT in the priming and
activation of monocytes by IFN-c, we have utilized specific
LPCAT inhibitors in an attempt to block this process. We
present herein evidence to show that LPCAT (but not other
acyltransferases) is a key regulator of both the priming of
monocytes by IFN-cand for LPS-initiated TNF-arelease.
Materials and methods
Materials
Unless stated otherwise, all reagents were from Sigma
Chemical Co (Poole, UK) and were of the best available
grades. Recombinant human IFN-cwas from Peprotech
(London, UK) and diluted in endotoxin-free water (Sigma).
Acyl-transferase inhibitors
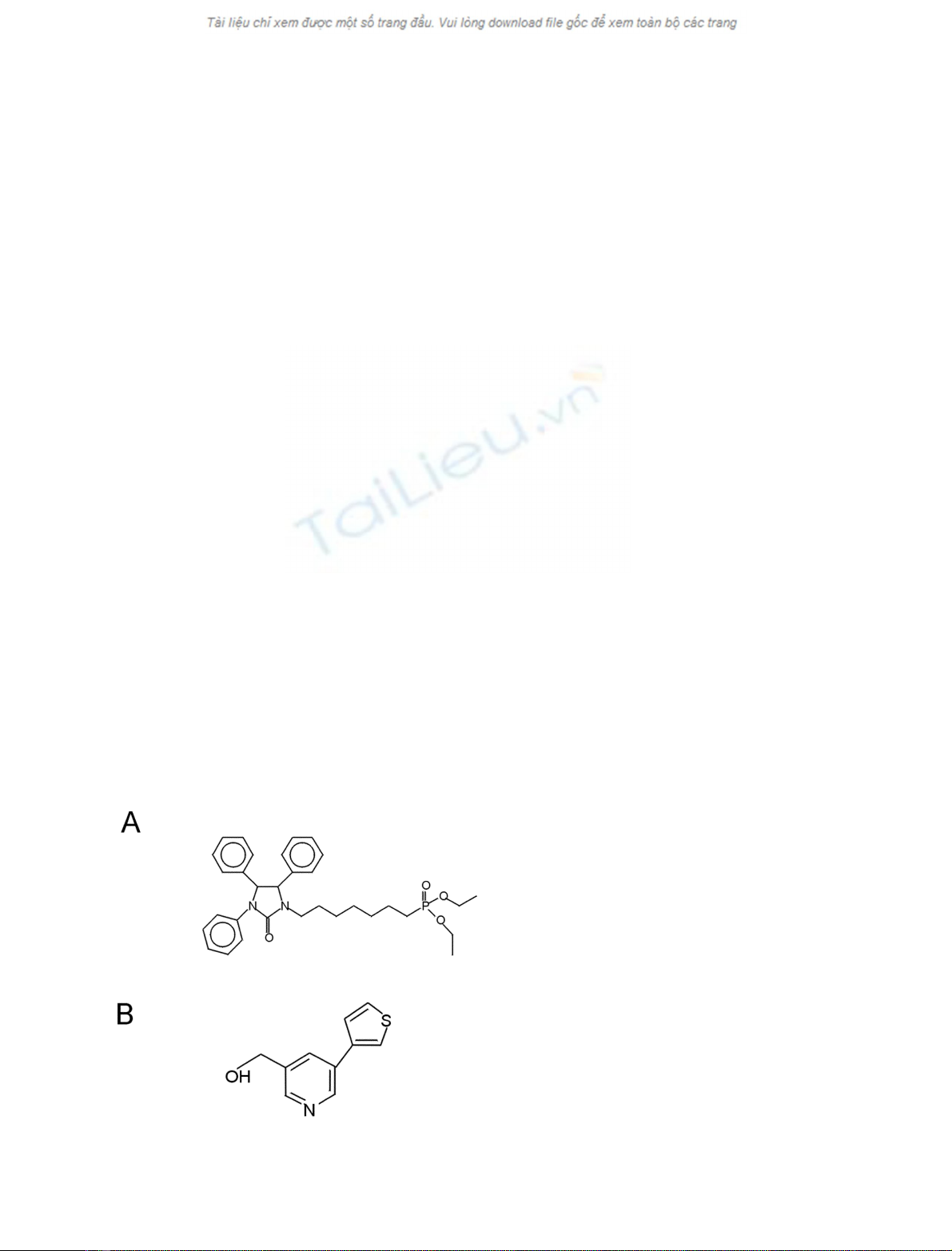
The acyltransferase inhibitor SK&F 98625 [diethyl 7-(3,4,
5-triphenyl-2-oxo2,3-dihydro-imidazole-1-yl)heptane phos-
phonate] [16] was purchased from Ferring Research Insti-
tute, Southampton, UK. YM 50201 (3-hydroxyethyl
5,3¢-thiophenyl pyridine), was a kind gift from the Yama-
nouchi Research Institute, Oxford, UK. Their structures are
given in Fig. 1. YM 50201 is a non-competitive inhibitor of
CoA-dependent LPCAT and has been shown to significantly
inhibit the incorporation of [
14
C]linoleic acid into lysophos-
phatidylcholine (M. J. Finnen, unpublished results)
1.
Cell culture
The acute monocytic leukemia cell line MonoMac-6 [17]
was obtained from the German Collection of Microorgan-
isms and Cell Cultures (Braunschweig, Germany). Cells
were grown in RPMI 1640 medium supplemented with 10%
(v/v) low endotoxin fetal bovine serum, 2 m
ML
-glutamine,
100 UÆmL
)1
penicillin, 100 UÆmL
)1
streptomycin, 1 m
M
sodium pyruvate, 1% (v/v) nonessential amino acids (all
from Life Technologies/Gibco, Paisley, UK) (complete
RPMI medium) and 8 lgÆmL
)1
bovine insulin (Sigma).
Cells were grown at 37 C and 5% (v/v) CO
2
in a humidified
incubator.
To isolate human monocytes, blood (10 mL) was collec-
ted from a healthy volunteer by venipuncture into a
heparinized tube and 2 mL Hypaque-Ficoll (Sigma) was
layered on top. The sample was centrifuged for 20 min at
400 gat room temperature and the mononuclear cell layer
removed to a fresh tube. The pellet was washed twice with
NaCl/P
i
. RPMI 1640 medium without additives was added
to cells to give a cell density of 5 ·10
6
mL
)1
.Toeachwell
of a 24-well plate, 400 lL of the cell suspension was added
and incubated at 37 C for 2 h. The monolayer was washed
thrice with warm RPMI 1640 medium. Fresh complete
RPMI 1640 medium (with all additives detailed above)
(1 mL) was added and the cells incubated at 37 Cas
indicated in the legends to the figures.
Cell viability was determined by dye exclusion. Equal
volumes of cell suspension and a 0.4% (v/v) solution of
trypan blue (Sigma) in 20 m
M
NaCl/Tris (pH 7.3) were
mixed. In all experiments viability was greater than 95%.
Preparation of microsomes
MonoMac-6 cells (5 ·10
6
) were incubated in 25 mL
complete RPMI 1640 medium under conditions indicated
in the legend to Fig. 2
2and harvested by centrifugation at
700 gfor 2 min at 4 C. The pellet was washed twice in
microsomal buffer (pH 7.4) containing sucrose 250 m
M
,
Tris 10 m
M
,MgCl
2
1m
M
,EGTA1m
M
[18]. The cells
were finally resuspended in 1 mL of microsomal buffer and
cells were lysed in an ice-cooled Dounce homogenizer
(2500 r.p.m., 2 min). Successful cell lysis was checked by
light microscopy. Another 1 mL of microsomal buffer was
added. To remove unbroken cells and large debris, the lysate
was centrifuged at 700 gfor 5 min and the resulting
supernatant was spun at 20 000 gfor 20 min and the final
supernatant at 100 000 gfor 1 h. The 100 000 gpellet thus
produced was resuspended in 1 mL microsomal buffer
(above)andstoredat)80 C. Protein concentration was
determined by the method of Bradford [19].
Measurement of coenzyme A-dependent acyltransferase
activity
Enzyme activity was determined by measuring the incor-
poration of radioactivity from acyl-CoA into diacyl-
phosphoglycerides. Aliquots (0.5 nmol; 0.7 kBq) of
[1–
14
C]oleoyl-CoA (Amersham) and 1 nmol 1-palmitoyl
lysophosphatidylcholine or 1-oleoyl lysophosphatidic acid
for each assay were dried under vacuum and resuspended in
75 lL assay buffer (150 m
M
NaCl, 1 m
M
EGTA and
10 m
M
Na
2
HPO
4
; pH 7.4) [18]. Microsomes (approxi-
mately 1 lgprotein)wereaddedin25lL assay buffer
and the mixture incubated with shaking for 20 min at
37 C. The reaction was stopped by the addition of 100 lL
Fig. 1. Structures of the acyltransferase inhibitors used. (A) SK&F
98625 [14] and (B) YM50201.
FEBS 2003 Monocyte acyltransferase in inflammation (Eur. J. Biochem. 270) 2783

chloroform/methanol (1 : 2, v/v). An additional 200 lL
chloroform, followed by 200 lL1
M
potassium chloride
were then added. After vortexing, the mixture was centri-
fuged at 400 gfor 2 min, the aqueous phase discarded and
the organic phase dried under vacuum at room temperature.
The latter was redissolved in 25 lL chloroform and applied
to a TLC plate (Silica gel 60, Merck, Darmstadt, Germany).
The samples were separated using a solvent of chloroform/
methanol/water (65 : 35 : 7, v/v/v). Authentic lipid stand-
ards (Avanti Polar Lipids, Alabaster, AL, USA) were
separated alongside the samples for identification. After
drying the plates, lipids were lightly stained with I
2
vapor
and their positions marked. Once the I
2
had evaporated,
individual lipid bands were scraped into scintillation vials
and radioactivity estimated in Optisafe Hisafescintillant
(Fisons, Loughborough, UK) using a LKB Wallac 1211
betacounter. Automatic quench correction was made by the
external standard channels ratio method. The data was
analyzed by Student’s unpaired t-test.
Measurement of TNF-a
MonoMac-6 cells (5 ·10
5
cells per well) were incubated in a
24-well tissue culture plate as detailed in the legend to Fig. 2.
3
Phorbol 12-myristate 13-acetate (PMA) (0.5 ngÆmL
)1
)was
added to the incubation medium to aid the secretion of
TNF-afrom the MonoMac-6 cells. At this concentration
PMA alone does not lead to a change in TNF-alevels [20].
Cells were harvested by centrifugation (1 min, 10 000 g,
room temperature) and nucleic acids extracted for nucleic
acid sequence based amplification (NASBATM)(seebelow).
The supernatants from the incubations were stored at
)80 C.
Production of TNF-aprotein by MonoMac-6 cells and
peripheral blood mononuclear cells was determined by an
ELISA method. All incubations were performed at room
temperature. An ELISA plate (Nunc Maxisorb) was loaded
with 100 lL antihuman TNF-amouse monoclonal capture
antibody (R&D, Minneapolis, MN, USA) in NaCl/P
i
(4 lgÆmL
)1
) per well and incubated overnight. The plate
was washed thrice with 0.05% v/v Tween 20 in NaCl/P
i
and
blotted dry. To reduce nonspecific binding, 300 lL bovine
serum albumin (BSA; 1%, w/v) and sucrose (5%, w/v) in
NaCl/P
i
were added to each well and the plates incubated
for at least 1 h and washed as above. Human recombinant
TNF-a(R&D) was diluted in dilution buffer (NaCl
150 m
M
, BSA 0.1%, Tween 20 0.05%, v/v, Tris 20 m
M
,
pH 7.3). Samples (supernatants from the incubations) were
diluted in the same buffer where appropriate. Standards
ranging from 15.6 to 1000 pgÆmL
)1
and samples (100 lL)
were added to the plate and incubated for 1 h. The plate
was washed as above and 100 lL200ngÆmL
)1
biotinylated
detection antibody (R&D, Abingdon, UK) in dilution
buffer added. After incubating for 2 h, the plate was
washed, 100 lL streptavidin–horseradish peroxidase conju-
gate (0.6 lgÆmL
)1
, Zymed, San Francisco, CA, USA) added
and the plate incubated for 20 min. The plate was washed
and 100 lL of tetramethylbenzidine substrate containing
0.01% H
2
O
2
(Kirkegaard and Perry, Gaithersburgh, MD,
USA) per well. The plate was incubated in the dark for
20 min and the reaction stopped by the addition of 50 lLof
0.5
M
sulfuric acid. The plate was read on a Labsystems
Multiskan plate reader using the absorbance difference,
A
450
)A
540
.
Detection of human TNF-amRNA by NASBATM
TNF-amRNA levels were measured by NASBATM (nucleic
acid sequence-based amplification) as described previously
[21]. Briefly, a silica-based extraction system was used to
prepare nucleic acids from MonoMac-6 cells. TNF-a
mRNA was amplified in the NASBATM process and
analyzed by polyacrylamide gel electrophoresis. Dried gels
were scanned on a Hewlett Packard photosmart scanner.
Results
Stimulation of monocyte acyltransferase activity by IFN-c
Our previous work [5,6] and that of others [22] has shown
that IFN-ccan profoundly alter the acyl chain compo-
sition of monocyte membrane phospholipids. Such alter-
ations could be attributed to Lands-type remodeling [23]
and were coincident with increased LPCAT activity [12].
To confirm that the modifications of phospholipids were
due to altered acyltransferase activity, the present study
measured the effect of IFN-con monocyte LPCAT and
LPAAT.
Acyltransferase activities were measured in microsomes
prepared from MonoMac-6 cells incubated in the presence
or absence of 250 UÆmL
)1
IFN-c. It has been shown
previously that this concentration of IFN-cmodulates the
response of monocytes to LPS [24]. The cytokine was found
to significantly increase LPCAT activity (P< 0.05), while
having no effect on LPAAT activity (Table 1). However,
LPS, added before or after IFN-c, did not affect the
LPCAT activity (data not shown). The stimulation of
LPCAT by IFN-ccould be inhibited by the tyrosine kinase
inhibitor tyrphostin (2.5 l
M
) but not by the protein kinase
C inhibitor bisindolylmalamide (1 l
M
) (data not shown).
This suggests that the signaling pathway from the IFN-c
receptor to the acyltransferase involves tyrosine kinases but
not protein kinase C. Such a finding is not unexpected as the
Janus family of cytoplasmic tyrosine kinases is known to
relay signals from IFN-creceptors to the nucleus, thereby
initiating gene activation [25].
Effect of priming on TNF-aproduction
IFN-cis well-known to up-regulate monocyte responses to
LPS [11,24] although the mechanism for this priming effect
is not well understood. We previously showed that mono-
cytes activated by IFN-cshowed phospholipid modifi-
cations consistent with increased LPCAT activation.
Therefore, in this study we wished to assess the effect of
blocking LPCAT activity on priming of monocytes by
IFN-c. To do this we utilized two acyltransferase inhibitors.
SK&F 98625, originally described as a CoA-independent
acyltransferase inhibitor [16], was included as it inhibits
both LPCAT and LPAAT in MonoMac 6 cells with IC
50
values of 10 l
M
and 30 l
M
, respectively (data not shown).
At 20 l
M
, the concentration used in our experiments, it
completely inhibits both LPCAT and CoA-independent
acyltransferase activity. In contrast, YM 50201 completely
2784 B. Schmid et al. (Eur. J. Biochem. 270)FEBS 2003


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)