
Determinants of antagonist binding at the a-amino-3-hydroxy-
5-methyl-4-isoxazolepropionic acid receptor subunit, GluR-D
Role of the conserved arginine 507 and glutamate 727 residues
Annukka Jouppila
1
, Olli T. Pentika¨ inen
2
, Luca Settimo
2
, Tommi Nyro¨ nen
3
, Jukka-Pekka Haapalahti
1
,
Milla Lampinen
1
, David G. Mottershead
1
, Mark S. Johnson
2
and Kari Keina¨ nen
1
1
Viikki Biocenter, Department of Biosciences (Division of Biochemistry) and Institute of Biotechnology, University of Helsinki,
Finland,
2
Department of Biochemistry and Pharmacy, A
˚bo Akademi University, Turku, Finland,
3
CSC – Scientific Computing Ltd,
Espoo, Finland., Subdivision in Eur. J. Biochem. Neurochemistry
Previous structural and mutagenesis studies indicate that the
invariant a-amino and a-carboxyl groups of glutamate
receptor agonists are engaged in polar interactions with
oppositely charged, conserved arginine and glutamate resi-
dues in the ligand-binding domain of a-amino-3-hydroxy-
5-methyl-4-isoxazolepropionic acid receptor. To examine
the role of these residues (R507 and E727 in the GluR-D
subunit) in the discrimination between agonists and anta-
gonists, we analyzed the ligand-binding properties of
homomeric GluR-D and its soluble ligand-binding domain
with mutations at these positions. Filter-binding assays using
[
3
H]AMPA, an agonist, and [
3
H]Ro 48–8587, a high-affinity
antagonist, as radioligands revealed that even a conservative
mutation at R507 (R507K) resulted in the complete loss of
both agonist and antagonist binding. In contrast, a negative
charge at position 727 was necessary for agonist binding,
whereas the isosteric mutation, E727Q, abolished all agonist
binding but retained high-affinity binding for [
3
H]Ro
48–8587, displaceable by 7,8-dinitroquinoxaline-2,3-dione.
Competition binding studies with antagonists representing
different structural classes in combination with ligand
docking experiments suggest that the role of E727 is anta-
gonist-specific, ranging from no interaction to weak elec-
trostatic interactions involving indirect and direct hydrogen
bonding with the antagonist molecule. These results under-
line the importance of ion pair interaction with E727 for
agonist activity and suggest that an interaction with R507,
but not with E727, is essential for antagonist binding.
Keywords: AMPA; ionotropic glutamate receptor; molecu-
lar modelling; Ro 48–8587; radioligand binding.
a-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
(AMPA)-selective glutamate receptors are multimeric
ligand-gated channels, which mediate fast excitatory
neurotransmission and, under pathological conditions,
contribute to the excitotoxic actions of glutamate [1]. As it
is believed that AMPA receptor antagonists may have
therapeutic potential as neuroprotective agents, detailed
information regarding the structural basis of ligand recog-
nition would be important for drug design and may also
help in the understanding of the activation mechanism of
the receptor [2].
AMPA receptors are assembled from a set of four
homologous subunit polypeptides, named GluR-A through
-D, or alternatively, as GluR1–4, each with 900 amino
acid residues and containing three predicted transmembrane
segments and a pore loop as part of the channel structure
[1,3]. Identification of homology between two segments (S1
and S2) in ionotropic glutamate receptor subunits and
bacterial amino acid-binding proteins [4,5], and expression
of a functional agonist binding site of the GluR-D and
GluR-B subunits of the AMPA receptor as soluble S1–S2
fusion proteins, [6,7] paved the way to the recent structure
determinations by Gouaux and coworkers, which provided
the first atomic resolution view of a neurotransmitter
binding site [8,9]. Crystal structures of S1–S2 constructs of
the GluR-B (GluR2) AMPA receptor subunit with bound
ligands show that the agonist ligands are buried deeply and
are engaged in multiple polar interactions with the two lobes
of the ligand-binding domain. The invariant a-aminocarb-
oxyl moiety of the agonists is stabilized through ion pair and
hydrogen bonding interactions involving the conserved
residues Arg485 in Lobe 1 and Glu705 in Lobe 2 as
predicted by site-directed mutagenesis [10]. (The numbering
of GluR-D residues is according to the virtual translation
Correspondence to K. Keina
¨nen, Department of Biosciences,
PO Box 56, Viikinkaari 5D, 00014 University of Helsinki,
Helsinki, Finland.
Fax: + 358 919159068, Tel.: + 358 919159606,
E-mail: kari.keinanen@helsinki.fi
Abbreviations:AMPA,a-amino-3-hydroxy-5-methyl-4-isoxazole-
propionic acid; ATOA, (RS)-2-amino-3-[5-tert-butyl-3-(carboxy-
methoxy)-4-isoxazolyl]propionic acid; ATPO, (RS)-2-amino-3-
[5-tert-butyl-3-(phosphonomethoxy)-4-isoxazolyl] propionic acid;
CNQX, 6-cyano-7-nitro-quinoxaline-2,3-dione; DNQX, 6,7-dinitro-
quinoxaline-2,3-dione; HEK, human embryonic kidney; PNQX,
(1,4,7,8,9,10-hexahydro-9-methyl-6-nitropyrido[3,4-f]quinoxaline-
2,3-dione); NS 257, (1,2,3,6,7,8-hexahydro-3-(hydroxyimino)-
N,N,7-trimethyl-2-oxobenzo[2,1-b:3,4-c¢] dipyrrole-5-sulfonamide);
NS 1209, 8-methyl-5-(4-(N,N-dimethylsulfamoyl)phenyl)-6,7,8,9,-
tetrahydro-1H-pyrrolo[3,2-h]-isoquinoline-2,3-dione-3-O-(4-hydroxy-
butyric acid-2-yl)oxime; Ro 48–8587, 9-imidazol-1-yl-8-nitro-2,3,5,6-
tetrahydro [1,2,4]triazolo[1,5-c] quinazoline-2,5-dione.
(Received 12 August 2002, revised 15 October 2002,
accepted 4 November 2002)
Eur. J. Biochem. 269, 6261–6270 (2002) FEBS 2002 doi:10.1046/j.1432-1033.2002.03345.x

from the corresponding nucleotide sequences starting with
the initiator methionine. This differs, by the length of the
predicted signal peptide, from the numbering used by
Gouaux and coworkers [8,9] for the mature GluR-B
(GluR2) polypeptide [8,9].) The three hydrogen bonds of
the a-amino group (to Pro478, Thr480, both in Lobe 1, and
Glu705) show a nearly perfect tetrahedral organization
around the central nitrogen. The distal acidic group of the
agonists interacts exlusively with Lobe 2 [9].
Importantly, the crystal structures of ligand-free GluR-B
S1–S2 and one antagonist complex, 6,7-dinitroquinoxaline-
2,3-dione (DNQX), have also been obtained [9]. These
complexes are slightly more open than the agonist com-
plexes, suggesting that agonist-induced closure of the lobes
may provide the driving force for channel opening [9].
Binding of the DNQX is mediated by polar contacts to both
lobes and aromatic stacking with the side chain of a tyrosine
residue. Consistent with pharmacophore models [11,12]
predicting that the carbonyl oxygens and iminic nitrogens of
the quioxalinedione antagonists, indispensable for activity,
mimic the a-aminocarboxylate core of the agonists, the
carbonyl oxygens of DNQX are hydrogen bonded to
Arg485. Glu705 does not, however, participate directly in
DNQX binding. Interestingly, however, this residue shows
two different conformations, one of which is pointing
directly below the quinoxalinedione ring and Glu705 is, in
fact, the only binding site residue which shows major side
chain reorientation between the agonist, antagonist and
ligand-free complexes [9].
Considering the large structural variation of AMPA
receptor antagonists [2,12], it can be expected that the
receptor–antagonist interactions may show differences to
those interactions seen in the DNQX complex structure. In
the present study, we have analyzed the ligand-binding
properties of mutated and wild-type homomeric GluR-D
AMPA receptors in order to further characterize the roles of
Arg507 and Glu727 (homologous to GluR-B residues
Arg485 and Glu705, respectively) in antagonist binding.
In order to separate the structural requirements necessary
for antagonist binding from those important for agonist
binding, we have used a high-affinity antagonist [
3
H]Ro
48-8587 [13] as a radioligand, in addition to [
3
H]AMPA.
MATERIALS AND METHODS
Experimental materials
CNQX, DNQX and kainic acid were obtained from RBI-
Sigma (Natick, MA, USA) and
L
-glutamate was from
Sigma (St Louis, MO, USA). [
3
H]AMPA (specific activity,
60 CiÆmmol
)1
) was from NEN Life Science Products
(Boston, MA, USA). [
3
H]Ro 48-8587 (specific activity,
44 CiÆmmol
)1
) was obtained from Amersham Pharmacia
Biotech (Buckinghamshire, UK). ATPO and ATOA [14]
were obtained from P. Krogsgaard-Larsen, Royal Danish
School of Pharmacy, Copenhagen, Denmark. NS 257 [15],
NS 1209 [16], and 1,4,7,8,9,10-hexahydro-9-methyl-6-nitro-
pyrido[3,4-f]quinoxaline-2,3-dione (PNQX) [11] were
obtained from J. Drejer (NeuroSearch A/S, Glostrup,
Denmark). Ro 48-8587 was obtained from R. Wyler and
V. Mutel (F. Hoffmann-La Roche, Basel, Switzerland).
Enzymes for molecular biology were purchased from New
England Biolabs (Beverly, MA, USA), Finnzymes (Espoo,
Finland), and Fermentas (Vilnius, Lithuania). Anti-Flag
M1 was obtained from Sigma. Anti-c-myc antibody was
purified from the hybridoma cell line 9E10.2 originally
obtained from American Type Culture Collection (ATCC
CRL1729).
DNA constructs and recombinant baculovirus
expression
Single point mutations were introduced into the flip isoform
[17] of N-terminally Flag-tagged or C-terminally myc-
tagged rat GluR-D in a pFASTBAC1 vector (BRL-Life
Sciences) [6,18] by overlap extension PCR using mutagenic
primers, followed by restriction fragment replacement. The
presence of the mutations was verified by DNA sequencing.
Recombinant baculovirus vectors were then prepared by
using the Bac-to-Bac system according to the manufac-
turer’s instuctions (Invitrogen Life Technologies, Carlsbad,
CA, USA), and used to infect monolayers of Trichoplusia ni
cells (High Five, Invitrogen) grown in T25 or T175 tissue
culture flasks at 27 C. SF900-II (Life Technologies, Paisley,
UK), supplemented with penicillin (100 lgÆmL
-1
), strepto-
mycin (lgÆmL
-1
) and amphotericin B (0.25 lgÆmL
-1
)was
used as the culture medium. The cells were collected 4 days
after infection by centrifugation (1000 g, for 10 min) and
analysed by immunoblotting. The cell pellets were stored at
)20 C until used for radioligand binding studies.
In some experiments, S1–S2 ligand-binding site con-
structs were used instead of the full-length receptor. The
Flag-tagged GluR-D S1–S2 constructs (wild-type, E727D
and E727A mutants), and their expression as soluble
proteins secreted in the culture medium have been described
previously [10].
Preparation of membranes
For radioligand binding experiments, membranes were
prepared as follows. Insect cells were homogenized in five
volumes of 20 m
M
Hepes, pH 7.4, 2.5 m
M
EDTA, 0.1 m
M
phenylmethanesulfonyl fluoride. The particulate fraction
was pelleted (30 000 g, 25 min) and washed extensively by
repeated homogenization and centrifugation in 20 m
M
Hepes, pH 7.4, 200 m
M
NaCl, 0.5 m
M
EDTA, 0.1 m
M
PMSF. Finally, the washed membranes were suspended in
20 m
M
Hepes, pH 7.4, 200 m
M
NaCl, 10% glycerol,
0.1 m
M
PMSF, and stored frozen or on ice until used in
the assay. Some experiments were performed with receptor
preparations that were solubilized in Triton X-100 as
described previously [19] with results essentially identical to
those obtained with membranes.
Radioligand binding assays
For the AMPA binding assays, the membranes were
suspended in 30 m
M
Tris/HCl, pH 7.2, 100 m
M
potassium
thiocyanate (KSCN), 2.5 m
M
CaCl
2
, 0.1% Triton X-100.
For initial measurement of binding activity and for ligand
competition experiments, 5 n
M
[
3
H]AMPA was used. For
saturation analyses, [
3
H]AMPAwasdilutedtoaspecific
activity of 12 CiÆmmol
-1
with unlabelled R,S-AMPA and
used at 1–300 n
M
concentrations. Membranes (20–250 lg
protein) were incubated in a 0.5-mL volume for 1 h on
ice with the radioligand, whereafter the reactions were
6262 A. Jouppila et al. (Eur. J. Biochem. 269)FEBS 2002

terminated by adding 5 mL of ice-cold 30 m
M
Tris/HCl,
pH 7.2, 100 m
M
KSCN, 2.5 m
M
CaCl
2
followed by rapid
filtration through Whatman GF/B filters with two 5-mL
washes with ice-cold buffer. Nonspecific binding was
determined in the presence of 1 m
ML
-glutamate. The filters
were solubilized in OptiPhase 3 HiSafe (Wallac) and
subjected to liquid scintillation counting.
[
3
H]Ro 48-8587 (0.5–2 n
M
final concentration) binding
assays were performed in 50 m
M
Tris/HCl, pH 7.0 [13]. The
filtration assay was performed as described for [
3
H]AMPA
binding with the exception that 50 m
M
Tris/HCl, pH 7.0
was used as the buffer. Saturation binding analysis with
[
3
H]Ro 48-8587 was performed by diluting the radioligand
with increasing amounts of unlabelled compound.
The ligand binding data were analyzed by nonlinear
curve fitting using the
PRISM
2.01 software (GraphPad Inc.).
Student’s t-test was used for the statistical analyses.
Molecular modelling
Structural modelling. The three-dimensional structures of
GluR-B S1–S2 ligand-binding core complexed with DNQX
(PDB accession no. 1ftl [9]); was obtained from the Protein
Data Bank [20]. The sequences of GluR-D and GluR-B
were aligned by using
MALIGN
[21] in the
BODIL
Modeling
Environment (Lehtonen J.V., Rantanen V.-V., Still, D.-J.,
Gyllenberg, M., and Johnson, M.S.; http://www.abo.fi/fak/
mnf/bkf/research/johnson/bodil.html, personal communi-
cation) using a structure-based sequence comparision
matrix [22]. The program
MODELLER
4.0 [23] was used to
construct a three-dimensional model structure by satisfying
spatial restraints imposed on the GluR-D sequence by its
alignment with the known GluR-B structure. At the same
time, the structure of the ligand seen in the X-ray structure
was built using
MODELLER
4.0.
Ligand minimization. Ligands were built with the program
SYBYL
6.6 (Tripos, St Louis, MO, USA) and energy
minimized prior to docking to the receptor models using
the
TRIPOS
Force Field and conjugate gradient method until
the energy gradient was less than 0.05 kcalÆmol
)1
. Protona-
tion of the polar groups was evaluated by comparison of the
final three-dimensional structures with similar substructures
obtained from the Cambridge Structural Data Bank (CSD
System Documentation, 1992, Cambridge Crystallographic
Data Centre, Cambridge, UK). To calculate the optimized
structure for Ro 48–8587, standard Hartree–Fock (HF/3-
21G) methods in
GAUSSIAN
98 [24] were used.
Receptor minimization. Polar hydrogens were added to the
receptor using
SYBYL
6.6. In order to optimize the
intramolecular interactions in the receptor model, hydrogen
atoms were minimized (keeping the rest of the model rigid)
using amber charges [25,26].
Ligand docking.
AUTODOCK
3.0 [27] is a semirigid docking
program that considers the whole ligand molecule docking
to the binding site and it is possible to choose the torsion
angles of the ligand that are allowed to rotate (AutoTors),
while the bond angles and bond lengths are kept fixed. The
overall interaction energy between chemical species is
estimated by considering both Lennard–Jones atom–atom
potentials and electrostatic effects, summed for the individ-
ual interactions between atoms. Partial charges for the
receptor model were calculated using the
AMBER
force field
in
SYBYL
6.6; and for ligands, the
MMFF
94 Force Field [28].
The interaction of a probe group (corresponding to each
type of atom in the ligand) with a receptor model was
calculated at grid positions 0.25 A
˚apart in a
(20 ·20 ·20) A
˚
3
box centered at the binding site using
the program
AUTOGRID
in
AUTODOCK
3.0. For each ligand,
10 separate docking simulations were performed.
Other assays. SDS/PAGE and Western blotting using
anti-Flag M1 (Sigma Chemical, St. Louis, MO, USA) as the
primary antibody, were performed as described previously
[6,19]. Protein content of the samples was measured by
using bicinchonic acid assay kit according to the manufac-
turer’s instructions. (BCA, Pierce, Rockford, IL, USA).
RESULTS
Effects of mutations at residues Arg507 and Glu727
on binding of [
3
H]AMPA to GluR-D
Structural and mutagenesis studies on soluble ligand-
binding domains indicate that Arg507 and Glu727 in the
AMPA receptor subunit GluR-D, and the equivalent
residues in the GluR-B subunit, serve as critical docking
sites for the a-amino and a-carboxylate groups of agonist
ligands. In the present study, we have examined the role of
these residues in antagonist recognition in the full-length,
membrane-bound AMPA receptor. Wild-type GluR-D and
its R507K, E727D and E727Q mutants were expressed in
recombinant baculovirus-infected Trichoplusia ni (High
Fig. 1. Expression and [
3
H]AMPA binding activity of GluR-D mutant
receptors. Wild-type and mutated epitope-tagged GluR-D AMPA
receptors were expressed in recombinant baculovirus-infected High
Five insect cells. (A) Immunoblot analysis showing the expression of
Flag-tagged GluR-D (WT), GluR-D E727D, GluR-D E727Q and of
myc-tagged GluR-D R507K. Noninfected cells were used as controls.
All lanes were loaded with an equal amount (10 lg) of membrane
protein, and the blots were developed by using anti-Flag M1 or anti-
myc 9E10 as primary antibodies as indicated. The positions of
molecular size markers are shown at the right. (B) Binding of
[
3
H]AMPA to mutant GluR-D receptors. Binding of 5 n
M
[
3
H]AMPA
to insect cell membranes (50 lg protein for wild-type and E727
mutants; 100 lg for R507K) was determined in the presence (non-
specific binding) and absence (total binding) of 1 m
M
glutamate.
Specific binding was defined as the difference between total and non-
specific binding. The values (mean ± SD) are from a representative
assay performed in triplicate.
FEBS 2002 Role of E727 of GluR-D in antagonist binding (Eur. J. Biochem. 269) 6263
Five) insect cells as homomeric, epitope-tagged receptors. In
immunoblots, an 105-kDa immunoreactive band, corres-
ponding to the size expected for a glycosylated GluR-D
monomer, was observed in baculovirus-infected cells ex-
pressing Flag-tagged GluR-D and the E727D and E727Q
mutants, and the myc-tagged R507K mutant, but not in
noninfected control cells (Fig. 1A). The presence of an
additional and intense 90-kDa myc-immunoreactive band
in cells expressing the R507K mutant suggests partial
proteolysis or defective glycosylation with this mutant
(Fig. 1A). To determine the ligand binding activity of the
GluR-D mutants, membrane preparations from the bacu-
lovirus-infected cells were subjected to a filtration binding
assay by using [
3
H]AMPA (at 5 n
M
) as the radioligand. In
accordance with a previous analysis conducted with the
soluble ligand-binding domain of GluR-D, [
3
H]AMPA
bound to wild-type GluR-D and to the E727D mutant but
not to the R507K and E727Q mutants (Fig. 1B). In a
saturation binding analysis, K
d
values of 40 and 11 n
M
were
obtained for the wild-type and E727D receptors, respect-
ively. The corresponding B
max
values were 12.3 pmolÆmg
)1
of protein for GluR-D and 14.7 pmolÆmg
-1
for GluR-D
E727D. In a competition binding assay, unlabelled
L
-glutamate exhibited a K
i
value of 0.20 ± 0.06 l
M
for
the wild-type GluR-D and 2.13 ± 0.13 l
M
for the GluR-D
E727D mutant (mean ± SD; n¼4). Kainate exhibited a
K
i
value of 2.10 ± 0.14 l
M
(mean ± SD; n¼4) with the
wild-type GluR-D, but displayed a drastically decreased
affinity to the E727D mutant (> 1000-fold), consistent with
earlier results obtained with the soluble ligand-binding
domain [10]. A reliable value for the inhibitory affinity
constant of kainate binding to E727D mutant receptor
could not be obtained, but 3 m
M
kainate caused only
20–25% inhibition of [
3
H]AMPA binding (not shown).
Interaction of competitive antagonists with wild-type
and E727D mutant receptors
Next, we determined the ability of competitive antagonists
representing different structural types (Fig. 2) to inhibit
[
3
H]AMPA binding to GluR-D and to the E727D mutant.
All eight antagonist ligands inhibited [
3
H]AMPA binding to
wild-type GluR-D in a concentration-dependent manner
(Fig. 3, Table 1). With the exception of two compounds,
Ro 48-8587 and NS 257, substituting an aspartate for the
glutamate at position 727 of GluR-D, produced significant
changes in the apparent binding affinitites of the antagonists
(Table 1). The closely related quioxaline-2,3-diones CNQX
and DNQX were (20-fold) more potent inhibitors of
[
3
H]AMPA binding at the mutant receptor, whereas an
opposite effect was seen with PNQX, NS 1209 and the two
AMPA-derivatives, ATPO and ATOA. In fact, the highest
concentration of ATOA which was tested (1 m
M
), did not
produce any clear inhibition of [
3
H]AMPA binding to the
E727D mutant. The competition curves were monophasic
and displayed Hill coefficients close to unity for all
antagonists, except for ATOA and ATPO whose low
affinity to the E727D mutant receptor prevented the
determination of more complete displacement curves.
Binding of [
3
H]Ro 48-8587 to wild-type and mutated
GluR-D receptors
As the E727Q and R507K mutant receptors were devoid of
any [
3
H]AMPA binding activity, the effect of these muta-
tions in antagonist recognition could not be studied by
binding inhibition experiments. Therefore, we employed a
recently introduced high-affinity antagonist radioligand,
[
3
H]Ro 48-8587 [13], to analyze directly the antagonist
binding properties of the mutant receptors. In a filtration
assay using a single 1 n
M
concentration of [
3
H]Ro 48-8587,
significant binding was observed to wild-type GluR-D, and
to the E727D and E727Q mutants (Fig. 4A). In contrast, no
binding above nonspecific background produced by mem-
branes prepared from noninfected cells was seen with the
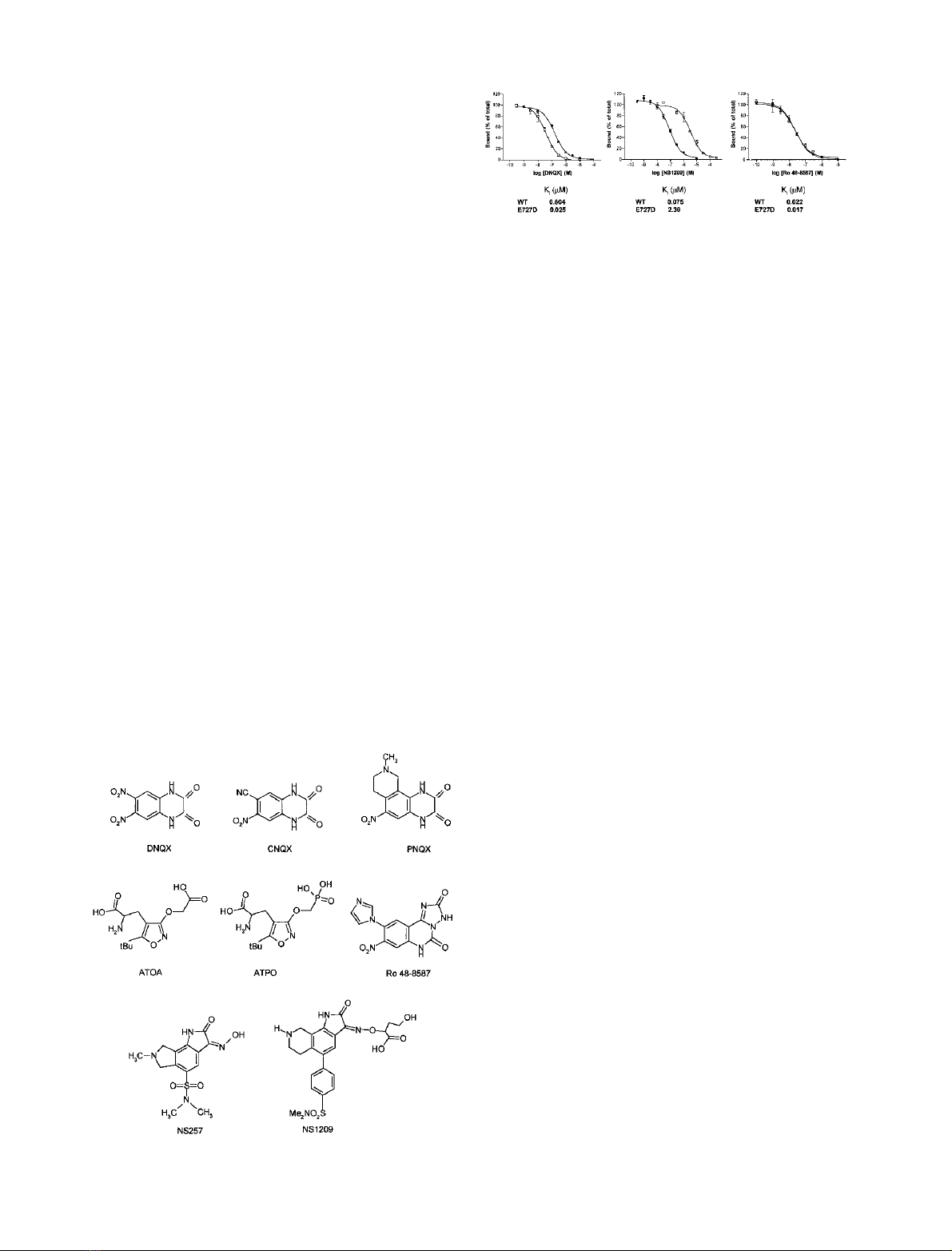
R507K mutant (Fig. 4A). In saturation binding experi-
ments, K
d
values of 8.3 ± 0.2 and 7.0 ± 1.2 n
M
were
obtained for the wild-type GluR-D and E727D, respect-
Fig. 2. Structures of AMPA receptor antagonists used in this study.
Fig. 3. Inhibition of [
3
H]AMPA binding by competitive antagonists.
Wild-type (solid squares) and E727D mutant (open squares) GluR-D
receptors were expressed in recombinant baculovirus-infected insect
cells. Insect cell membranes were equilibrated with [
3
H]AMPA (5 n
M
)
in the presence of increasing concentrations of unlabelled antagonist
compounds, and the amount of bound [
3
H]AMPA was determined by
rapid filtration. Displacement of [
3
H]AMPA binding is shown for
DNQX, NS 1209, and Ro 48–8587. The curves represent a best fit to a
one-site model obtained by nonlinear curve fitting.
6264 A. Jouppila et al. (Eur. J. Biochem. 269)FEBS 2002

ively, whereas a slightly higher K
d
value, 20.0 ± 0.5 n
M
,
was obtained for E727Q (mean ± SD; n¼3; Fig. 4B).
In order to further substantiate the findings and to
facilitate comparision with earlier work, we performed some
[
3
H]Ro 48-8587 binding studies with the separately
expressed S1–S2 ligand-binding domain of GluR-D
(Fig. 4C). Consistent with the results obtained with the
membrane-bound full-length receptors, [
3
H]Ro 48-8587 (at
2n
M
concentration) bound specifically to the wild-type and
E727D S1–S2 proteins, whereas the S1–S2 R507K mutant
did not show binding activity (Fig. 4D). In a saturation
binding analysis, K
d
values of 15.6 ± 6.6 and
13.0 ± 1.8 n
M
were obtained for the binding of
[
3
H]Ro 48-8587 to wild-type S1–S2 and to the E727D
mutant, respectively (mean ± SD; n¼3). Furthermore, an
S1–S2 protein carrying E727A mutation, previously shown
to lack [
3
H]AMPA binding acitivity ([10,] Fig. 4D), did not
show any [
3
H]Ro 48-8587 binding in the filtration assay
(Fig. 4D).
Ligand-binding properties of E727Q mutant receptors
Because earlier mutagenesis studies indicated that a negative
charge at position 727 is necessary for agonist binding, the
ability of the E727Q mutant receptor to bind [
3
H]Ro
48-8587 was of considerable interest, and therefore we
examined further the [
3
H]Ro 48-8587 binding properties of
homomeric E727Q receptors by using ligand competition
experiments. Due to the limited amount of [
3
H]Ro 48-8587
available, only semiquantitative analyses were performed.
Using a single 1 m
M
concentration of unlabelled ligand, the
agonists,
L
-glutamate and kainate, were practically inactive
as inhibitors of [
3
H]Ro 48-8587 binding to the E727Q
receptor, whereas DNQX inhibited > 90% of the binding
(Fig. 5A). As expected, all three ligands strongly inhibited
[
3
H]Ro 48-8587 binding to wild-type GluR-D, whereas
kainate was a significantly weaker inhibitor than glutamate
and DNQX at E727D membranes, consistent with the
dramatic decrease in kainate affinity caused by E727D
mutation (Fig. 5A). Furthermore, DNQX and NS 1209
inhibited [
3
H]Ro 48-8587 binding to GluR-D E727Q
membranes with K
i
values of 0.033 ± 0.011 and
2.52 ± 0.15 l
M
, respectively (mean ± SD; n¼3;
Fig. 5B), indicating that binding of these two antagonists
(as with Ro 48–8587) can bind to the receptor in the
presence of glutamine at position 727. Unfortunately, the
small amount of labelled [
3
H]Ro 48-8587 available preven-
ted more extensive displacement experiments.
Molecular model of GluR-D ligand-binding domain
Ligand binding studies were complemented by docking
simulations in order to help us understand the role of
R507 and E727 in the interactions of the receptor with
the different antagonists. First, a three-dimensional
model of the ligand binding domain of GluR-D (S1–
S2) was built using the 1.8 A
˚resolution structure of the
GluR-B S1–S2-DNQX complex [9] as a template. The
sequence identity between the molecules was high
(90%) with only one difference located near the
binding site, Tyr703 in GluR-B being replaced by a
phenylalanine in GluR-D.
Ligand docking
Quinoxalinediones. DNQX was docked automatically
into the GluR-D receptor model resulting in very similar
interactions to those present in the crystal structure of
GluR-B S1–S2-DNQX complex, including formation of
two hydrogen bonds with R507 (Fig. 6A). Glu727 does
not form hydrogen bonds with DNQX and is likely to
have the same two side chain orientations as the
equivalent Glu705 has in the asymmetric unit of GluR-
B S1–S2-DNQX complex. Both orientations could ac-
commodate a weak electrostatic interaction with the
DNQX ring structure, which carries a partial positive
charge due to two strongly electron-withdrawing nitro
groups. Furthermore, in the GluR-B S1–S2-DNQX
complex [9], Tyr732 is seen to form a hydrogen bond
with the 6-nitro group of DNQX or with Glu705,
depending on the orientation of the side chain of
Glu705. Similar interactions are likely to take place
between DNQX and the corresponding residues, Glu727
and Tyr754, in GluR-D (Fig. 7A and B).
Table 1. Inhibition of [
3
H]AMPA binding to GluR-D and GluR-D E727D by competitive antagonists. Wild-type and E727D mutant GluR-D
receptors were expressed in recombinant baculovirus-infected insect cells. Insect cell membranes were equilibrated with 5 n
M
[
3
H]AMPA in the
presence of increasing concentrations of the indicated unlabelled antagonists. Non-specific binding, determined in the presence of 1 m
M
glutamate,
was subtracted from all values. The specific K
i
values were calculated by using the Cheng–Prusoff equation with K
d
values of 40 and 11 n
M
for
[
3
H]AMPA binding to the wild-type receptor and E727D, respectively. The values represent the mean ± SD from three to five independent
determinations (ngiven in parenthesis). Statistical significance of the difference between the wild-type GluR-D and E727D mutant was determined
by using Student’s t-test and is indicated as P-values. ND, not determined.
Antagonist GluRD GluRD E727D P-value
CNQX 0.433 ± 0.167 (3) 0.022 ± 0.007 (4) 0.0038
DNQX 0.546 ± 0.180 (4) 0.016 ± 0.006 (4) 0.0011
PNQX 0.334 ± 0.105 (5) 2.57 ± 1.30 (4) 0.0060
NS 257 0.834 ± 0.110 (5) 0.682 ± 0.092 (5) 0.0464
NS 1209 0.087 ± 0.035 (4) 1.61 ± 0.68 (4) 0.0042
ATOA 437 ± 118 (3) > 1000
a
(3) ND
ATPO 67 ± 6 (3) > 1000
b
(3) ND
Ro 48–8587 0.020 ± 0.004 (4) 0.022 ± 0.003 (4) 0.454
a
No inhibition was obtained at the highest concentration used: 97.7 ± 4.1% of the binding left at 1 m
M
ATOA.
b
Only a partial inhibition
was obtained: 65.3 ± 3.5% of the binding left at 1 m
M
ATPO.
FEBS 2002 Role of E727 of GluR-D in antagonist binding (Eur. J. Biochem. 269) 6265


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)