
Role of histidine 42 in ascorbate peroxidase
Kinetic analysis of the H42A and H42E variants
Latesh Lad
1
, Martin Mewies
1
, Jaswir Basran
2
, Nigel S. Scrutton
2
and Emma L. Raven
1
1
Department of Chemistry and
2
Department of Biochemistry, University of Leicester, UK
To examine the role of the distal His42 residue in the cata-
lytic mechanism of pea cytosolic ascorbate peroxidase, two
site-directed variants were prepared in which His42 was
replaced with alanine (H42A) or glutamic acid (H42E).
Electronic spectra of the ferric derivatives of H42A and
H42E (pH 7.0, l¼0.10
M
,25.0C) revealed wavelength
maxima [k
max
(nm):397,509,540
sh
, 644 (H42A); 404, 516,
538
sh
, 639 (H42E)] consistent with a predominantly five-
co-ordinate high-spin iron. The specific activity of H42E for
oxidation of
L
-ascorbate (8.2 ± 0.3 UÆmg
)1
)was30-fold
lower than that of the recombinant wild-type enzyme
(rAPX); the H42A variant was essentially inactive but
activity could be partially recovered by addition of exogen-
ous imidazoles. The spectra of the Compound I intermedi-
ates of H42A [k
max
(nm) ¼403, 534, 575
sh
, 645] and H42E
[k
max
(nm) ¼404, 530, 573
sh
, 654] were similar to those of
rAPX. Pre-steady-state data for formation of Compound I
for H42A and H42E were consistent with a mechanism
involving accumulation of a transient enzyme intermediate
(K
d
) followed by conversion of this intermediate into
Compound I (k¢
1
). Values for k¢
1
and K
d
were, respectively,
4.3 ± 0.2 s
)1
and 30 ± 2.0 m
M
(H42A) and 28 ± 1.0 s
)1
and 0.09 ± 0.01 m
M
(H42E). Photodiode array experi-
ments for H42A revealed wavelength maxima for this
intermediate at 401 nm, 522 nm and 643 nm, consistent
with the formation of a transient [H42A–H
2
O
2
] species. Rate
constants for Compound I formation for H42A were
independent of pH, but for rAPX and H42E were pH-
dependent [pK
a
¼4.9 ± 0.1 (rAPX) and pK
a
¼6.7 ± 0.2
(H42E)]. The results provide: (a) evidence that His42 is
critical for Compound I formation in APX; (b) confirmation
that titration of His42 controls Compound I formation and
anassignmentofthepK
a
for this group; (c) mechanistic and
spectroscopic evidence for an intermediate before Com-
pound I formation; (d) evidence that a glutamic acid residue
at position 42 can act as the acid–base catalyst in ascorbate
peroxidase.
Keywords: ascorbate peroxidase; Compound I; histidine 42.
The plant peroxidase superfamily has been classified [1] into
three major categories: class I contains the enzymes of
prokaryotic origin, class II contains the fungal enzymes (e.g.
manganese peroxidase, lignin peroxidase) and class III
contains the classical secretory peroxidases [e.g. horseradish
peroxidase (HRP)]. The most notable member of the class I
peroxidase subgroup is cytochrome cperoxidase (CcP),
which was first identified in 1940 [2]. In spite of the fact that
CcP has some rather unusual features, most notably the
existence of a stable tryptophan radical during catalysis
[3–6] and the utilization of a large macromolecular substrate
(cytochrome c), it has been the subject of such intense
mechanistic, structural and spectroscopic scrutiny that it has
become the benchmark against which all other peroxidases
are measured.
More recently, it has been possible to isolate and purify in
good yields a second member of the class I peroxidase
subgroup, ascorbate peroxidase (APX) [7,8]. Ascorbate-
dependent peroxidase activity was first reported in 1979
[9,10] and the enzyme catalyses the reduction of potentially
damaging H
2
O
2
in plants and algae using ascorbate as a
source of reducing equivalents [11,12]. APX was known
from sequence comparisons [13] to contain the same active-
site Trp residue (Trp179) as is used by CcP (Trp191) during
catalysis. With high-resolution structural information avail-
able for the recombinant pea cytosolic enzyme (rAPX) [14]
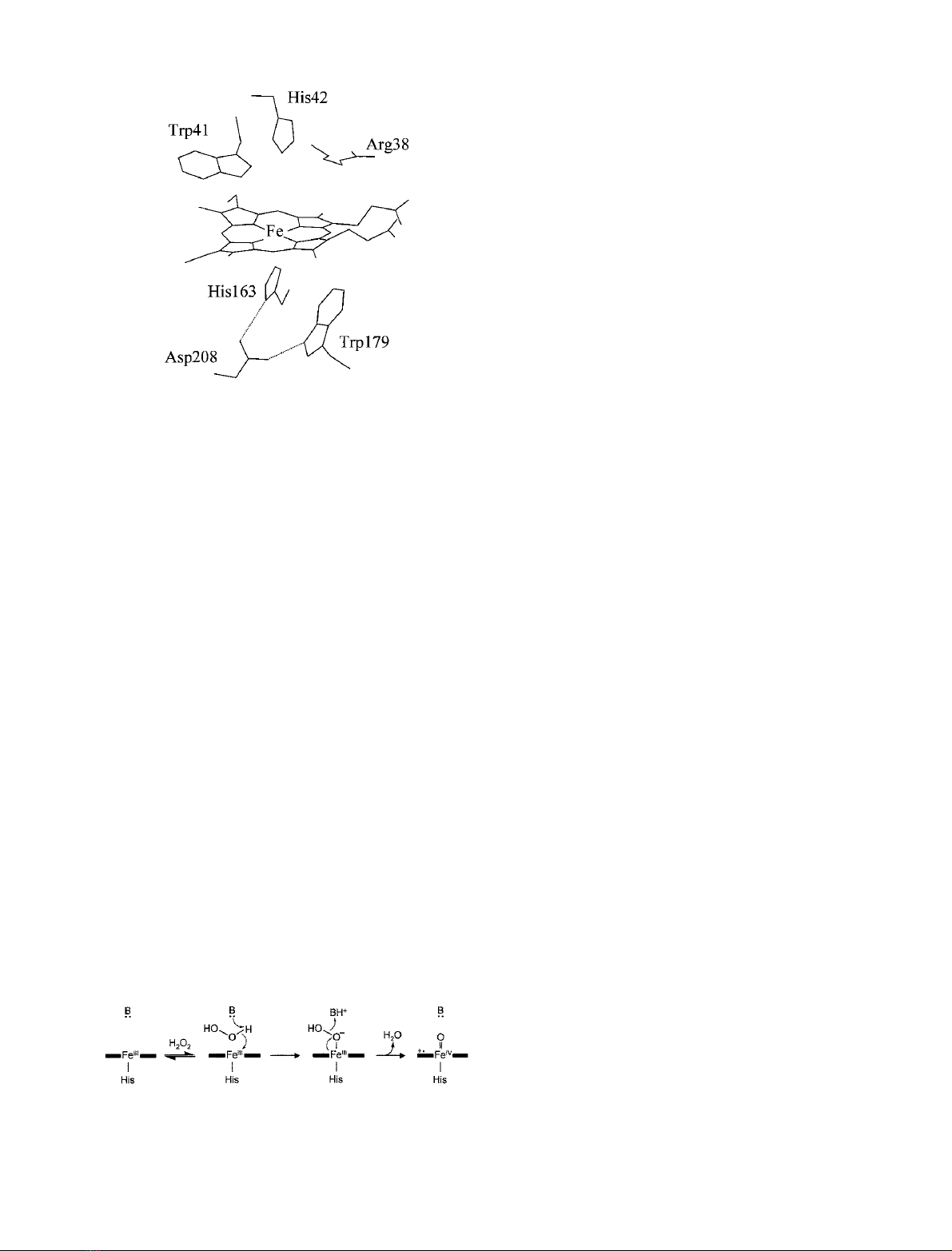
(Fig. 1), APX has provided a new opportunity to reassess
the functional properties of CcP and to determine whether it
is indeed representative of class I peroxidases. As detailed
functional information has emerged, however, it seems that
APX has several rather curious features of its own, and, in
some ways, more questions have been raised than answered.
(In fact, even the current classification of APX as a class I
enzyme has been recently questioned [15].) For example,
Trp179 in APX is not a necessary requirement for oxidation
of ascorbate [16] and there is general agreement from kinetic
[17–19] and EPR data [20] that the initial product
(Compound I) of the reaction of APX with H
2
O
2
is a
porphyrin p-cation intermediate and not a protein-based
trytophan radical. Equally intriguing is the existence of a
Correspondence to E. L. Raven, Department of Chemistry,
University of Leicester, University Road, Leicester LE1 7RH, UK.
Fax: + 44 (0)116 2523789, Tel.: + 44 (0)116 2522099,
E-mail: emma.raven@le.ac.uk
Abbreviations: APX, ascorbate peroxidase; pAPX, wild-type pea
cytosolic APX; rAPX, recombinant wild-type pea cytosolic APX;
H42A, a variant of rAPX in which His42 has been replaced
with alanine; H42E, a variant of rAPX in which His42 has been
replaced with glutamic acid; CcP, cytochrome cperoxidase; HRP,
horseradish peroxidase;
sh
, shoulder.
Enzymes: ascorbate peroxidase (EC 1.11.1.11); horseradish peroxidase
(EC 1.11.1.7); cytochrome cperoxidase (EC 1.11.1.5).
(Received 27 December 2001, revised 8 April 2002,
accepted 9 May 2002)
Eur. J. Biochem. 269, 3182–3192 (2002) FEBS 2002 doi:10.1046/j.1432-1033.2002.02998.x
potassium-binding site (not present in CcP), located 8A
˚
from the a-carbon of Trp179; the functional role of this site
(if, indeed, there is one) is not yet fully understood.
Although steady-state kinetic analyses have been a fairly
prominent feature of most of the early literature on APX
(reviewed in [12]), pre-steady-state kinetic data have been
very much more limited, largely as a result of insufficient
quantities of enzyme, and only preliminary mechanistic
information is available [16–19,21,22]. The enzyme operates
through a classical peroxidase mechanism in which the ferric
enzyme is oxidized by two electrons to a so-called
Compound I intermediate with concomitant release of
one molecule of water, followed by two successive single-
electron reductions of the intermediate by ascorbate (HS) to
regenerate ferric enzyme.
APX þH2O2!
k1Compound I þH2Oð1Þ
Compound I þHS !
k2Compound II þSð2Þ
Compound II þHS !
k3APX þSþH2Oð3Þ
Although Eqn (1) is commonly written as a single step,
experimental [23–26] and theoretical [27–30] evidence sug-
gests that this is an over-simplification. A more complex
mechanism, as first suggested by Poulos and Kraut [31] –
involving binding of neutral peroxide to the enzyme,
concomitant proton transfer from the bound peroxide to
the distal histidine residue, followed by O–O bond cleavage
and release of H
2
O – has been suggested (Scheme 1). Of
particular interest is the role of the distal histidine residue
(His42 in APX), which has been proposed [31,32] to act as
an acid–base catalyst, by accepting a proton from H
2
O
2
and
releasing it subsequently as water. Site-directed mutagenesis
studies on CcP and HRP have provided evidence to support
these predictions (reviewed in [33–37]). In the work reported
here, we replaced the distal histidine residue of APX (Fig. 1)
with alanine and glutamic acid (H42A and H42E variants,
respectively). The aims of the work were severalfold. First,
to establish a definitive role for His42 in Compound I
formation by replacing it with a residue that is not capable
of hydrogen bonding (H42A) and to examine whether other
residues at this position are able to act as alternative acid–
base catalysts (H42E). Secondly, to use these variants to
provide information on the origin of the pH-dependent
kinetic rate profile for Compound I formation [17]. Finally,
as we anticipated that the replacement of His42 would
probably generate variant enzymes that may well have
altered kinetic properties, we sought to utilize these alter-
ations in intimate mechanism to probe in more detail the
formation of Compound I in APX. As such, we present the
first spectroscopic evidence for the nature of the interme-
diate formed during the reaction of APX with H
2
O
2
.
MATERIALS AND METHODS
Materials
L
-Ascorbic acid (Aldrich Chemical Co.), guaiacol, imida-
zole, 1,2-dimethylimidazole (Sigma Chemical Co.) and the
chemicals used for buffers (Fisher) were of the highest
analytical grade (more than 99% purity) and used without
further purification. H
2
O
2
solutions were freshly prepared
by dilution of a 30% (v/v) solution (BDH): exact concen-
trations were determined using the published absorption
coefficient (e
240
¼39.4
M
)1
Æcm
)1
[38]). Aqueous solutions
were prepared using water purified through an Elgastat
Option 2 water purifier, which itself was fed with deionized
water. All pH measurements were made using a Russell pH-
electrode attached to a digital pH-meter (Radiometer
Copenhagen, model PHM 93).
Mutagenesis and protein purification
Site-directed mutagenesis was performed according to the
QuikchangeTM protocol (Stratagene Ltd, Cambridge, UK).
Two complementary oligonucleotides encoding the desired
mutation were synthesized and purified (PerkinElmer). For
H42A, the primers were: 5¢-CGTTTGGCATGGGCT
TCTGCTGGTAC-3¢(forward primer) and 3¢-GCAAAC
CGTACCCGAAGACGACCATG-5¢(reverse primer). For
H42E the primers were: 5¢-CGTTTGGCATGGGAATC
TGCTGGTAC-3¢(forward primer) and 3¢-GCAAACC
GTACCCTTAGACGACCATG-5¢(reverse primer). To
confirm the identity of the transformants, overnight cultures
containing 100 lgÆmL
)1
ampicillin were incubated at 37 C
with vigorous shaking (250 r.p.m.). The plasmid DNA was
isolated using the Hybaid mini-plasmid system and
sequenced to confirm the desired mutation. Automated
fluorescent sequencing, using New England Biolabs pUC
and malE primers, was performed by the Protein and
Nucleic Acid Chemistry Laboratory, University of Leices-
ter, on an Applied Biosystems 373-Stretch machine, and
Scheme 1. Proposed steps in the formation of Compound I. The
mechanism depicts the neutral peroxide-bound and anionic peroxide-
bound intermediates. The distal histidine residue that acts as the acid–
base catalyst is indicated (B).
Fig. 1. Active site of ascorbate peroxidase [14]. Hydrogen bonds
(dotted lines) are indicated.
FEBS 2002 Catalytic mechanism of ascorbate peroxidase (Eur. J. Biochem. 269) 3183

sequence data were analysed using the program SeqED
(Applied Biosystems). Individual mutations were confirmed
by sequencing across the whole rAPX-coding gene.
Bacterial fermentation of cells and purification of rAPX
were carried out according to published procedures [7].
Enzyme purity was assessed by examination of the A
Soret
/
A
280
value; in all cases an A
Soret
/A
280
value > 1.9 for rAPX,
H42A and H42E was considered pure. Enzyme purity was
additionally assessed using SDS/PAGE, and the prepara-
tions were judged to be homogeneous by the observation of
a single band on a Coomassie Blue-stained reducing SDS/
polyacrylamide gel. Enzyme concentrations (pH 7.0,
l¼0.10
M
, 25.0 C) were determined using the pyridine
haemochromagen method [39]: absorption coefficients were
e
403
¼88 m
M
)1
Æcm
)1
for rAPX [40], e
397
¼83 m
M
)1
Æcm
)1
for H42A and e
404
¼95 m
M
)1
Æcm
)1
for H42E.
UV/visible spectroscopy
Spectra were recorded using a variable-slit Perkin-Elmer
Lambda 14 UV/visible spectrometer, linked to an Exacta
486D computer, and an Epsom-LQ-1060 printer. Tempera-
ture was controlled (± 0.1 C) using a thermally jacketed
cell holder connected to a circulating water bath (Julabo
U3) and a water cooler (MK Refrigeration Limited), which
was operated in tandem.
Mass spectrometry
Samples were analysed using a Micromass Quattro BQ
(Tandem Quadrupole) electrospray mass spectrometer.
Horse heart myoglobin (Sigma) was prepared as described
for rAPX below, and used to calibrate the spectrometer in
the range 600–1400 m/z. Protein samples were introduced
into the instrument at a flow rate of 5 lLÆmin
)1
. Trace salt
was removed using a Centricon-10 concentrator (Amicon)
and successive centrifugation and dilution with highly
purified water (Elgastat). Samples (2mgÆmL
)1
,20lL)
were then diluted 10-fold with a solution of 50 : 50 (v/v)
acetonitrile/water containing 0.1% acetic acid.
Steady-state measurements
Stock solutions of
L
-ascorbic acid, guaiacol, H
2
O
2
and
enzyme were prepared in sodium phosphate (l¼0.10
M
,
pH 7.0, 25.0 C). Enzyme assays were performed in a 1-mL
quartz cuvette: various concentrations of substrate and
25 n
M
enzyme were preincubated for 3 min in buffer and
the reaction was initiated by the addition of H
2
O
2
(2.5 lL, 30 m
M
) to a final concentration of 0.1 m
M
.
The wavelengths and absorption coefficients used for
various substrates were as follows:
L
-ascorbic acid,
e
290
¼2.8 m
M
)1
Æcm
)1
[41]; guaiacol, e
470
¼22.6 m
M
)1
Æcm
)1
[42]. Activities were determined by dividing the change in
absorbance by the absorption coefficient of the substrate.
Values for k
cat
were calculated by dividing the maximum
rate of activity (l
M
)1
Æs
)1
) by the micromolar concentration
of enzyme; values for K
m
were determined by a fit of the
data to the Michaelis–Menten equation using a nonlinear
regression analysis program (Grafit32 version 3.09b;
Erithacus Software Ltd). All reported values are the
mean of three independent assays. Errors on k
cat
and K
m
are estimated to ± 5% and ± 10%, respectively. For
pH-dependent assays, a mixed sulfonic acid buffer system
(l¼95–110 m
M
depending on the exact pH) that buffered
over the entire pH range was used; reactions were initiated
by the addition of H
2
O
2
(to 0.10 m
M
). In these cases,
[
L
-ascorbic acid] ¼0.70 m
M
(rAPX) and 0.50 m
M
(H42E),
[guaiacol] ¼30 m
M
(rAPX) and 11 m
M
(H42E), and
[enzyme] ¼25 n
M
. Specific activities ([enzyme] ¼25 n
M
,
sodium phosphate, pH 7.0, l¼0.10
M
,25.0C) were
calculated from initial slopes of activity measurements;
1 unit of activity is defined as the amount of enzyme that
oxidizes 1 lmol substrate per minute (lmolÆmin
)1
Æmg
)1
).
Transient-state kinetics
Transient-state kinetics were performed using a SX.18 MV
microvolume stopped-flow spectrophotometer (Applied
Photophysics) fitted with a Neslab RTE200 circulating
water bath (± 0.1 C). Reported values of k
obs
are an
average of at least three measurements. All curve fitting was
performed using the Grafit software package. All data were
analysed using nonlinear least-squares regression analysis
on an Archimedes 410–1 microcomputer using Spectra-
kinetics software (Applied Photophysics). Pseudo-first-
order rate constants for the formation of Compound I
(k
1,obs
) were monitored at 403 nm (rAPX), 404 nm (H42E)
and 397 nm (H42A), in single mixing mode by mixing
enzyme (0.5–1.0 l
M
) with various concentrations of H
2
O
2
.
Absorbance changes were independent of [H
2
O
2
]; observed
changes in absorbance were 97–99% of the calculated
values. The pH-jump method was used to examine the
pH-dependence of Compound I formation, to avoid enzyme
instability problems below pH 5 and above pH 8.5. Enzyme
samples were prepared in water, adjusted to pH 7 with trace
amounts of phosphate buffer (5 m
M
,pH8.0);H
2
O
2
solutions were made up in buffers of twice the final
concentration. The buffers used were sodium phosphate in
the pH range 5.5–8.5 (l¼0.20
M
), citrate-phosphate in the
pH range 4.0–6.0 (l¼0.20
M
) and carbonate buffer in the
range 8.0–9.0 (l¼0.20
M
). The pH of the solution was
measured after mixing to ensure consistency. pH-dependent
data were fitted to the Henderson–Hasselbach equation for
a single-proton process (Eqn 4):
k¼AþB10pHpKa
1þ10pHpKað4Þ
where Aand Bare the rate constants for Compound I
formation at the extremes of acidic and basic pH, respect-
ively, and kis the rate constant (either second-order, k
1
,for
rAPX or limiting first-order, k¢
1
, for H42A/H42E) for
Compound I formation. Formation of Compound I in the
presence of exogenous imidazole was carried out using
single-wavelength mode (397 nm), where one syringe con-
tained H42A (1 l
M
)andtheotherH
2
O
2
(0.5–35 m
M
)inthe
presence of either imidazole or 1,2-dimethylimidazole
(20 m
M
) (relatively low concentrations of exogenous imi-
dazole and a high buffer concentration were used to
minimize the effect of fluctuating imidazole levels on the
ionic strength and pH and to prevent binding of
the imidazole to the haem). Time-dependent spectra of the
various reactions were obtained by multiple-wavelength
stopped-flow spectroscopy using a photodiode array detec-
tor and
X
-
SCAN
software (Applied Photophysics). Spectral
3184 L. Lad et al.(Eur. J. Biochem. 269)FEBS 2002
deconvolution was performed by global analysis and
numerical integration methods using
PROKIN
software
(Applied Photophysics).
RESULTS
Mass spectrometry
The integrity of the variant proteins was examined to ensure
that post-translational modification of the protein had not
occurred. Analysis of H42A and H42E (data not shown)
gave average masses for the apoproteins of 27126.9 ±
0.8 Da and 27185.1 ± 0.5 Da, respectively, in good agree-
ment with the calculated masses of 27126.74 Da (H42A)
and 27184.88 Da (H42E).
Electronic spectra
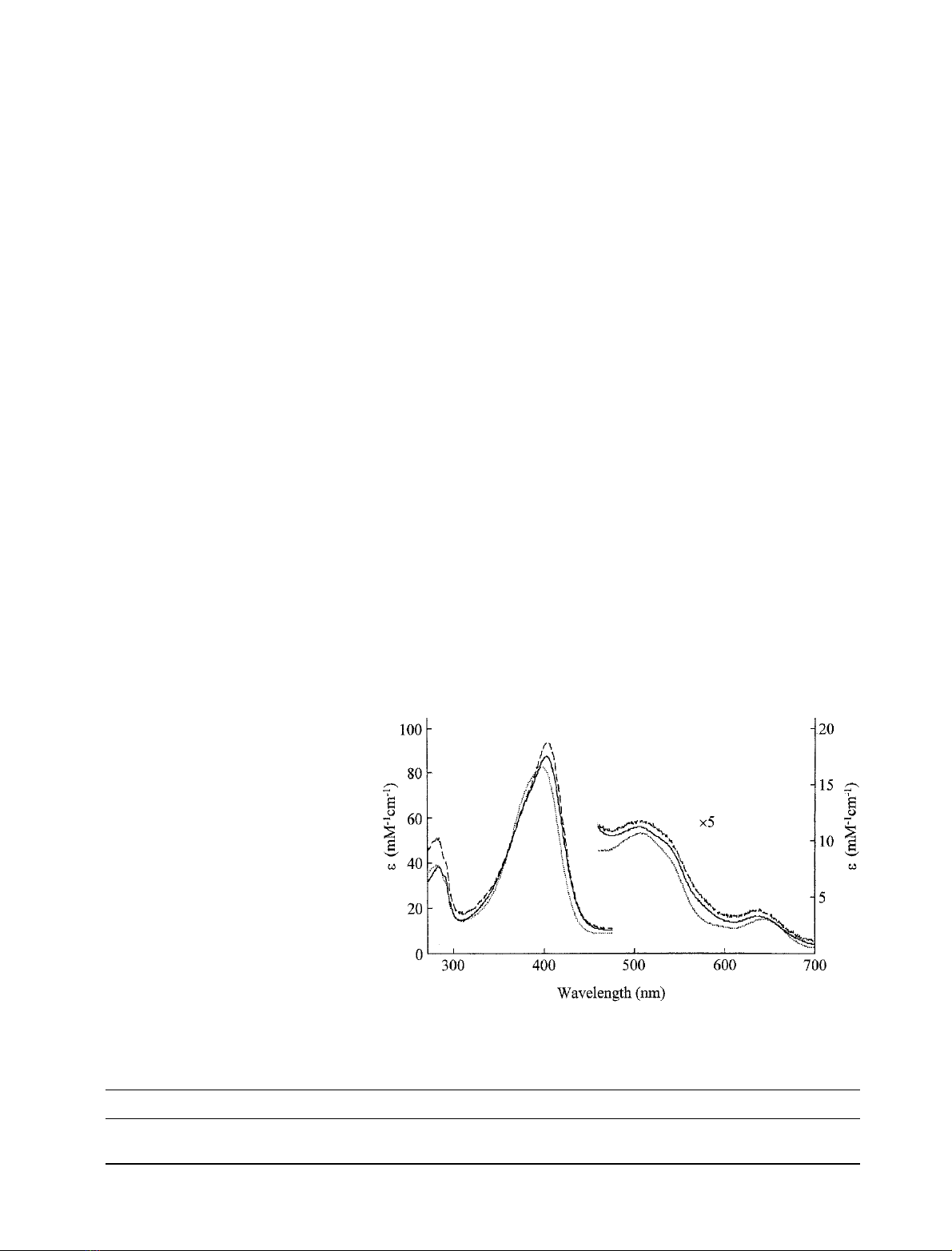
Electronic spectra of the ferric derivatives of H42A and
H42E (pH 7.0, l¼0.10
M
, 25.0 C)areshowninFig.2.
Wavelength maxima (Table 1) for H42A and H42E were
found to be slightly different from those for rAPX, but are
consistent with a predominantly five-coordinate high-spin
iron. In the presence of excess cyanide, spectra for H42A
and H42E were consistent with the formation of a low-spin
haem species, with absorption maxima [H42A–CN k
max
(nm) (e(m
M
)1
Æcm
)1
)) ¼420 (102), 540, 574
sh
; H42E–CN
k
max
(nm) (e(m
M
)1
Æcm
)1
)) ¼420 (109), 540, 573
sh
] similar
to those for the corresponding derivative of rAPX [rAPX–
CN k
max
(nm) (e(m
M
)1
Æcm
)1
)) ¼419 (104), 539, 572
sh
]. The
spectra of both ferric H42A and H42E were, on the other
hand, unaffected by the addition of either azide or fluoride,
suggesting that these (weak field) ligands do not bind to the
haem under these conditions.
Steady-state kinetics
The specific activity of rAPX for oxidation of
L
-ascorbic
acid (256 ± 6 UÆmg
)1
) is comparable to published data
(411 UÆmg
)1
) for pAPX [8]. The specific activity of H42E
(8.2 ± 0.3 UÆmg
)1
)was30-fold lower than that of
rAPX. Under conditions identical with those used for
rAPX and H42E, H42A exhibited no activity with
L
-ascorbic acid, although residual activity was detected
(9.2 ± 0.3 ·10
)2
UÆmg
)1
) when a higher enzyme concen-
tration was used ([H42A] ¼200 n
M
).
Steady-state data (k
cat
,K
m
and the arithmetically calcu-
lated selectivity coefficient, k
cat
/K
m
) for oxidation of
L
-ascorbic acid and guaiacol by rAPX and H42E are shown
in Table 2. (The oxidation of
L
-ascorbic acid by rAPX does
not obey standard Michaelis kinetics [8,22,43] and, in this
case, data were fitted to the Hill equation. Oxidation of
guaiacol by rAPX obeys Michaelis kinetics and data were
fitted to the Michaelis–Menten equation. The origin of the
different concentration-dependencies for these two sub-
strates is not known. Oxidation of both
L
-ascorbic acid and
guaiacol by H42E was observed to obey Michaelis–Menten
kinetics.) For both substrates, k
cat
values for H42E are 50-
fold lower than for rAPX, with K
m
values largely unaffected
(about threefold lower for H42E). The H42A variant was
inactive with both
L
-ascorbic acid (above) and guaiacol. The
dependence of the rate of substrate oxidation (l
M
Æs
)1
)vs.
pH yielded a pH optimum for rAPX at 7forthe
oxidation of both
L
-ascorbic acid (pH optimum 7.0) and
guaiacol (pH optimum 6.9) (data not shown) (the pH
optimum for
L
-ascorbic acid is consistent with that reported
previously for pAPX [8]). For H42E, the pH optimum is
shifted by 1 pH unit for both
L
-ascorbic acid (pH
optimum 8.1) and guaiacol (pH optimum 8.0).
Fig. 2. UV/visible spectra of ferric rAPX (solid
line), H42A (dotted line) and H42E (dashed
line). The region 450–700 nm has been multi-
plied by a factor of five. Sample conditions:
sodium phosphate, pH 7.0, l¼0.10
M
,
25.0 C.
Table 1. Wavelength maxima (nm) and in parentheses absorption coefficients (m
M
)1
Æcm
)1
) for the ferric and Compound I derivatives of rAPX, H42A
and H42E.
Derivative rAPX H42A H42E
Fe
III
403(88), 506, 540
sh
, 636 397(83), 509, 540, 644 404(95), 516, 538
sh
, 639
Compound I 404(59), 529, 583
sh
, 650 403(62), 534, 575
sh
, 645 404(66), 530, 573
sh
, 654
FEBS 2002 Catalytic mechanism of ascorbate peroxidase (Eur. J. Biochem. 269) 3185
Pre-steady-state kinetics
Spectra of the transient Compound I intermediates of
H42A and H42E, formed by reaction of the ferric deriva-
tives with 10 equivalents of H
2
O
2
, were obtained by
photodiode array experiments. These preliminary experi-
ments showed that Compound I formation for both
variants is much slower than for rAPX, reactions being
complete in less than 1000 s for H42A and 100 s for H42E,
compared with less than 300 ms for rAPX under identical
conditions. Wavelength maxima and absorption coefficients
for the Compound I intermediate of H42A and H42E were
found to be similar to those of rAPX (Table 1) and to those
previously published for wild-type pAPX (k
max
¼404 nm
[17]). For H42A and H42E, Compound I is surprisingly
stable, for up to 30 s, but does not spontaneously convert
into Compound II as is observed for rAPX. Instead,
Compound I for both H42A and H42E slowly returns to
a spectrum with a slightly red-shifted Soret band, with
wavelength maxima at 405, 514, 550
sh
and 640 nm for
H42A and 406, 516, 552
sh
and 638 nm for H42E.
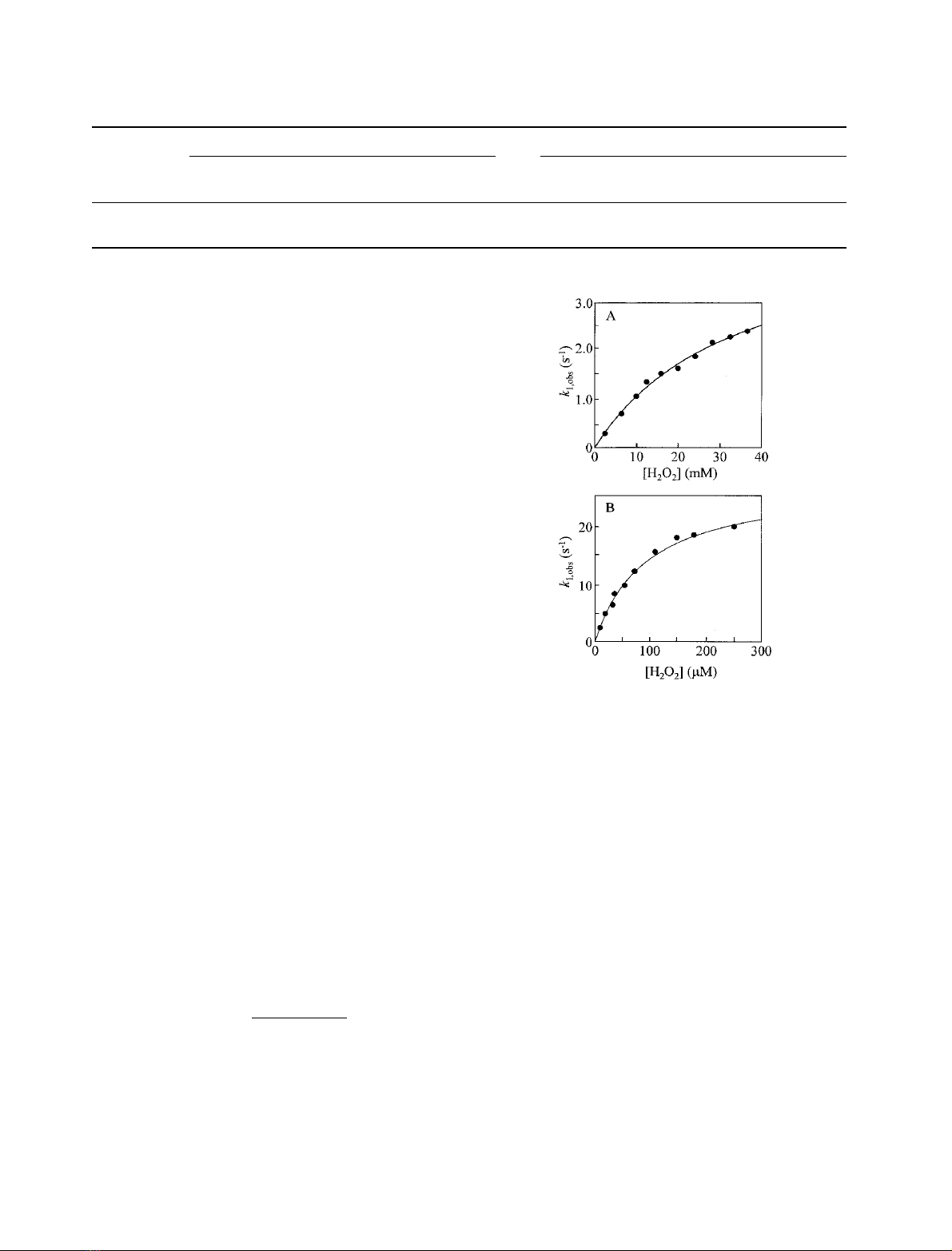
The dependence of the observed rate constant, k
1,obs
(individual traces were monophasic in all cases), on the
concentration of H
2
O
2
for H42A and H42E (Fig. 3)
exhibits hyperbolic behaviour. For rAPX (data not shown
and [21,22]) and pAPX [17], a linear dependence on [H
2
O
2
]
is observed; in this work, a second-order rate constant of
(6.1 ± 0.1) ·10
7
M
)1
Æs
)1
was derived for reaction of rAPX
with H
2
O
2
. Saturation behaviour of the kind exhibited by
H42A and H42E is consistent with a mechanism involving a
pre-equilibrium step which precedes Compound I forma-
tion (Eqns 5 and 6):
EþH2O2*
)
KaXð5Þ
X!
k0
1Compound I þH2Oð6Þ
(E ¼H42A or H42E). This mechanism predicts a linear
(first-order) dependence at low concentrations of peroxide
and a zero-order dependence at high concentrations. An
expression for k
1,obs
can be derived (Eqn 7):
k1;obs ¼k0
1
1þKd=½H2O2ð7Þ
where K
d
is the dissociation constant of the bound complex
in Eqn (5) (K
d
¼1/K
a
)andk¢
1
is the limiting first-order rate
constant at high peroxide concentrations. A fit of these data
for H42A and H42E to Eqn (7) (Fig. 3) yields values for k¢
1
and K
d
of 4.3 ± 0.2 s
)1
and 30 ± 2.0 m
M
, respectively
(H42A) and 28 ± 1.0 s
)1
and 0.09 ± 0.01 m
M
, respect-
ively (H42E). These data pass through the origin, indicating
that the second step of the reaction is irreversible. At low
concentrations of peroxide, where a linear dependence is
observed, it is possible to extract an approximate value for
the second-order rate constant for reaction with H
2
O
2
[Eqn
(5) where K
d
is related to the microscopic second-order (k
a
)
and first-order (k
b
) rate constants for this step (K
d
¼k
b
/
k
a
)]: values for k
a
of 84 ± 6
M
)1
Æs
)1
and 1.1 ± 0.2 ·
10
5
M
)1
Æs
)1
were obtained for H42A and H42E, respectively.
Themechanisticschemeimplicatedbytheabovedata
suggested the accumulation of a reaction intermediate, the
conversion of which to product was rate-limiting at high
peroxide concentrations. To examine the nature of this
intermediate, photodiode array experiments were carried
out for H42A (Fig. 4). Intermediate spectra were obtained
from a spectrally deconvoluted model: A fiBfiC,
where A corresponds to ferric H42A, B corresponds to the
intermediate (tentatively assigned as the [H42A–H
2
O
2
]
complex, vide infra) and C corresponds to Compound I.
Wavelength maxima for the proposed intermediate were at
401 nm, 522 nm and 643 nm. The model yielded rate
constants for each step: k
A
(A fiB) and k
B
(B fiC) of
Fig. 3. Dependence of k
1,obs
on [H
2
O
2
] concentration for the reaction of
H42A (A) and H42E (B) with H
2
O
2
(sodium phosphate, pH 7.0
l¼0.10
M
,5.0°C, [H42A] ¼0.5 l
M
,[H42A]¼0.5 l
M
). Data were
fitted using a nonlinear least-squares fitting procedure to Eqn (7).
Table 2. Values for k
cat
,K
m
and k
cat
/K
m
for the oxidation of
L
-ascorbic acid and guaiacol by rAPX and H42E (sodium phosphate, pH 7.0,
l¼0.10
M
).
L
-Ascorbate Guaiacol
Enzyme k
cat
(s
)1
)K
m
(m
M
)
k
cat
/K
m
(m
M
)1
Æs
)1
)k
cat
(s
)1
)K
m
(m
M
)
k
cat
/K
m
(m
M
)1
Æs
)1
)
rAPX 248 ± 28 0.41 ± 0.04 605 66 ± 3 12.3 ± 0.9 5.4
H42E 3.9 ± 0.2 0.17 ± 0.09 22.9 1.5 ± 0.1 2.9 ± 0.35 0.5
3186 L. Lad et al.(Eur. J. Biochem. 269)FEBS 2002


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)