
N-
myc
oncogene overexpression down-regulates leukemia inhibitory
factor in neuroblastoma
Elissavet Hatzi
1
, Carol Murphy
1,2
, Andreas Zoephel
3
, Horst Ahorn,
3
Ulrike Tontsch
3
, Ana-Maria Bamberger
4
,
Keiko Yamauchi-Takihara
5
, Lothar Schweigerer
6
and Theodore Fotsis
1
1
Laboratory of Biological Chemistry, Medical School, University of Ioannina, Greece;
2
Biomedical Research Institute,
Ioannina, Greece;
3
Boehringer Ingelheim Austria GmbH, Vienna, Austria;
4
Institute of Pathology, Department of Gynecophathology,
University Hospital Hamburg Eppendorf, Hamburg, Germany;
5
Department of Molecular Medicine, Osaka University Graduate
School of Medicine, Suita, Japan;
6
Abt. Ha
¨matologie, Onkologie und Endokrinologie, Universita
¨ts-Kinderklinik Essen, Germany
Amplification of N-myc oncogene is a frequent event in
advanced stages of human neuroblastoma and correlates
with poor prognosis and enhanced neovascularization.
Angiogenesis is an indispensable prerequisite for the pro-
gression and metastasis of solid malignancies, which is
modulated by tumor suppressors and oncogenes. We have
addressed the possibility that N-myc oncogene might regu-
late angiogenesis in neuroblastoma. Here, we report that
experimental N-Myc overexpression results in down-regu-
lation of leukemia inhibitory factor (LIF), a modulator of
endothelial cell proliferation. Reporter assays using the LIF
promoter and a series of N-Myc mutants clearly demon-
strated that down-regulation of the LIF promoter was
independent of Myc/Max interaction and required a
contiguous N-terminal N-Myc domain. STAT3, a down-
stream signal transducer, was essential for LIF activity as
infection with adenoviruses expressing a phosphorylation-
deficient STAT3 mutant rendered endothelial cells insensi-
tive to the antiproliferative action of LIF. LIF did not
influence neuroblastoma cell proliferation suggesting that, at
least in the context of neuroblastoma, LIF is involved in
paracrine rather than autocrine interactions. Our data shed
light on the mechanisms by which N-myc oncogene ampli-
fication enhances the malignant phenotype in neuroblas-
toma.
Keywords:N-myc; LIF; STAT3; endothelial cell; neuro-
blastoma.
The N-myc proto-oncogene encodes a 64-kDa nucleopro-
tein (N-Myc) which associates with a 21- to 22-kDa Max
protein to form N-Myc/Max heterodimers [1]. These dimers
can bind to the E-box consensus sequence (CACGTG) in
the promoter regions of target genes [2], including alpha
prothymosin and ornithine decarboxylase, eventually
inducing their up-regulation [3]. The physiological functions
of N-Myc have remained elusive although there is evidence
for developmentally important activities [4–6]. In the neural
crest, enhanced N-Myc expression may facilitate prolifera-
tion of immature neuronal precursor cells at the expense of
differentiation [7,8].
N-myc is implicated in the pathogenesis of neural crest-
derived tumors including neuroblastoma [9], the most
frequent solid malignancy of infants. Amplification of
N-myc oncogene is a frequent event in advanced stages (III
and IV) of human neuroblastoma [10] and correlates with
poor prognosis [11]. In fact, N-myc amplification is nearly
exclusively observed in neuroblastoma. N-myc amplifica-
tion results in high N-Myc protein levels that could perturb
the finely tuned interplay of N-Myc and Max and eventually
induce abnormal expression patterns of target genes [1].
However, few N-Myc target genes have been identified so
far [1]. A number of data indicate that some of the target
genes might modulate the cardiovascular system. Indeed,
whereas N-myc knockout mice die in utero [12,13], com-
pound heterozygotes suffer from serious heart defects [6].
Moreover, N-myc may also regulate the growth of tumor
vessels as neuroblastoma with N-myc amplification exhibit
enhanced neovascularization [14] suggesting that N-Myc
oncogene could stimulate tumor angiogenesis and thereby
enhance neuroblastoma progression. Indeed, stable trans-
fection and 100-fold overexpression of N-Myc in a neuro-
blastoma cell line (SH-EP) resulted in an enhanced
malignant phenotype of the transfectants (WAC2) and the
ability to form well vascularized tumors in nude mice [15].
As tumor angiogenesis derives from an imbalance
between angiogenic factors and inhibitors [16,17], it is
conceivable that the genetic changes of cancer could initiate
angiogenesis by disturbing this balance in the tumor vicinity.
Indeed, normal p53 regulates the expression of the angio-
genesis inhibitors thrombospondin [18] and glioma-derived
angiogenesis inhibitory factor [19]. Also, activation of
oncogenes, such as ras, has been shown to cause both
up-regulation of the expression of angiogenesis stimulators,
such as basic fibroblast growth factor (bFGF) or vascular
endothelial growth factor (VEGF), and down-regulation of
angiogenesis inhibitors such as tissue inhibitor of matrix
metalloproteinases (TIMP) and thrombospondin [20].
Towards the aim of identifying N-myc-regulated mole-
cules modulating neuroblastoma tumor angiogenesis, we
Correspondence to T. Fotsis, Laboratory of Biological Chemistry,
Medical School, University of Ioannina, 45110 Ioannina, Greece.
Tel.: + 30 65197560, Fax: + 30 65197868,
E-mail: thfotsis@cc.uoi.gr
Abbreviations: LIF, leukemia inhibitory factor; bFGF, basic fibroblast
growth factor; VEGF, vascular endothelial growth factor; TIMP,
tissue inhibitor of matrix metalloproteinases.
(Received 20 February 2002, revised 30 May 2002,
accepted 21 June 2002)
Eur. J. Biochem. 269, 3732–3741 (2002) FEBS 2002 doi:10.1046/j.1432-1033.2002.03066.x

have previously screened conditioned media from SH-EP
control transfectants (SH-EP007) and WAC2 cells for the
presence of inhibitors or stimulators of endothelial cell
proliferation [21]. We were able to demonstrate that three
endothelial cell proliferation inhibitors present in SH-EP007
supernatants (SI.1, SI.2, and SI.3) were completely down-
regulated in WAC2 cells [21]. In a further study, we have
identified SI.3 as being activin A and documented antian-
giogenic properties for this TGF-family member [22]. The
present study deals with the structural and functional
characterization of SI.1.
MATERIALS AND METHODS
Cell culture and cell proliferation assays
Dishes, media and recombinant growth factors have been
described previously [21,23]. Cells were cultured as described
[15,23,24]. Collection of conditioned medium from
SH-EP007 cells was carried out as mentioned before [21].
Cell proliferation assays using BBCE cells were previously
described [21,23]. Briefly, cells were seeded (day 0) in 12-well
tissue culture plates at a density of 1250 cellsÆcm
)2
(5000 cells per well) and the following day (day 1), wells
received 10 lL of the fractions to be tested and 2.5 ngÆmL
)1
bFGF. This treatment was repeated after two days (day 3).
On day 5 or 6, cells in duplicate wells were trypsinized and
counted using a Coulter particle counter. SHEP-007 and
WAC2 cells were seeded at a density of 2500 cellsÆcm
)2
(10 000 cells per well). For growth curves of WAC2 stable
transfectants, cells were seeded in 12-well tissue culture
plates at a density of 2500 cellsÆcm
)2
(10 000 cells per well)
and counted daily using a Coulter particle counter. Goat
anti-human polyclonal LIF neutralizing antibody was
obtained from R & D systems.
Purification of SI.1
Concentration of conditioned medium (47 L), acidification
and SP-Sepharose Fast Flow chromatography were carried
out as described previously [21]. Fractions containing the
SI.1 were subjected to concavalin A affinity chromatogra-
phy. A concavalin A–Sepharose column was used (11-mL
bed volume, Pharmacia) equilibrated in 50 m
M
Tris/HCl,
pH 7.2 containing 500 m
M
NaCl, 0.1% Chaps, 1 m
M
CaCl
2
and 1 m
M
MnCl
2
. Samples were applied in equil-
ibration buffer, and elution was carried out using: (a) 10 bed
volumes of equilibration buffer; (b) a linear gradient
consisting of 5 bed volumes of equilibration buffer and 5
bed volumes of equilibration buffer containing 500 m
M
a-
methyl mannopyranoside; and (c) 5 bed volumes of
equilibration buffer containing 500 m
M
a-methyl manno-
pyranoside. A flow rate of 12 cmÆh
)1
was used and fractions
of 2.5 mL were collected.
The active fractions were desalted, lyophilized and
subjected to a preparative SDS/PAGE electrophoresis in
tubes (150 ·5 mm) containing 80 ·5 mm of resolving
(12%) and 30 ·5 mm of stacking (4%) polyacrylamide gel.
The tubes and buffers were according to Laemmli [21a] and
electrophoresis was carried out with buffer recirculation at
2 mA per tube (1 W maximun power). Following electro-
phoresis the tubes were washed for 30 min in 1% Chaps in
NaCl/P
i
followed by a further 30 min wash in NaCl/P
i
.
After exchange of detergent from the denaturing SDS to the
nondenaturing Chaps, the polyacrylamide tubes were cut in
2.5 mm slices and a small piece of each slice was allowed to
diffuse, at 4 Covernight,in700lL of endothelial cell
medium. The diffusates and/or their dilutions were tested
for inhibitory activity on endothelial cell proliferation.
The slices corresponding to the active fractions were then
lyophilized and the bound proteins were allowed to difuse to
tissue culture tested water. Following concentration, they
dissolved in 0.25% trifluoroacetic acid/distilled H
2
Oand
loaded onto a Bakerbond Wide-Pore C
18
RP-HPLC)
column (4.6 ·250 mm; Malinckrodt Baker, Philipsburg,
NJ, USA) equilibrated with 0.1% trifluoroacetic acid.
Bound material was eluted with a linear gradient from 0
to 30% acetonitrile in 15 min and 30–60% acetonitrile in
45 min at a flow rate of 1 mLÆmin
)1
. Fractions of 1 mL
were collected, lyophilized and dissolved in tissue culture
water for activity evaluation on the proliferation of
endothelial cells and amino-acid sequencing.
Mass fingerprint analysis and microsequencing
The active fractions from the HPLC step were electropho-
resed on a 12.5% SDS-polyacrylamide gel under nonreduc-
ing conditions and stained with Coomassie blue R250.
Protein bands were excised from the stained gel, destained,
reduced and alkylated with iodoacetamide, and digested with
trypsin. The resulting peptides were subjected to mass
spectroscopy analysis using a MALDI-TOF instrument
(Voyager DE-STR, PerSeptive Biosystems). Peptide samples
were prepared using dihydroxybenzoic acid as matrix. From
the calibrated MS peptide mass map a peak table list of the
peptide-mass fingerprint were designed omitting signals
observed in the chemical background spectrum. The peak
table list served as input data and the
MS
-
FIT
software
program (K. Clauser & P. Baker, available from http://
prospector.ucsf.edu/ucsfhtml4.0/msfit.htm) was used for
searching the SWISS Prot and NCBI protein databases for
sequence similarities and thus suggesting the identity of the
protein. Parameters were set for mass tolerance at 50 p.p.m.,
minimum number of peptides required at 4, molecular mass
of proteins from 1000 to 10 000 Da, protein pI from 3 to 10,
cysteines modified as amidomthylated.
MS
-
FIT
search results
were checked regarding the MOWSE score, molcular mass
(Da), pI, species and % masses matched. Edman sequencing
of the RP-HPLC separated tryptic peptides was performed
using an 494 cLC ABI-PerkinElmer apparatus.
Transfections and LIF promoter reporter assays
Full-length N-myc (pcDNA3-N-myc) was generated by PCR
amplification using pN-myc as template (a gift from M.
Schwab, German Cancer Research Center, Heidelberg,
Germany) [24]. All deletion mutants of N-myc were gener-
ated from the above construct by ligation of PCR products.
All constructs were sequenced. Transient transfections were
performed using lipofectamine-plus transfection reagent
(Invitrogen) according to the manufacturer’s instructions.
A reporter luciferase construct containing the 666 bp
human LIF promoter fragment (phLIF-Luc) was used [25].
SH-EP007 cells were plated at a density of 5 ·10
5
cells per
well in a six-well plate were transfected with 0.4 lLofthe
phLIF-Luc construct, alone or in combination with 0.4 lgof
FEBS 2002 N-myc oncogene down-regulates LIF (Eur. J. Biochem. 269) 3733

wild-type or mutated N-myc expression vectors for 28 h. In
each transfection a constant total DNA concentration was
used. Cells were collected and analyzed for luciferase activity
using a kit according to the manufacturer (Promega,
Madison, WI, USA) and a luminometer (EG & G Junior).
As internal control of transfection efficiency, 0.2 lgof
b-galactosidase expression vector (CMV-b-gal) was cotrans-
fected and the enzymatic activity was measured. The
expression and nuclear localization of all constructs was
determined by Western blot using a mouse antihuman
N-myc antibody (0.3 lgÆmL
)1
, Cymbus Biotechnology Ltd).
The full-length cDNA encoding human LIF, kindly
donated by Y. Jacques (INSERM U211, Nantes, France),
was subcloned into the XhoIsiteofaCMVpromoter
construct. WAC2 cells (70% confluent) were transfected
with effectene (Qiagen) using 1 lg of the LIF construct and
0.1 lg of a construct containing puromycin resistance gene
(Clontech). Clones were selected in medium containing
0.7 lgÆmL
)1
puromycin and expression of LIF was evalu-
ated by Western blot analysis of supernatants using a goat
anti-LIF Ig (R & D). Several independently derived clones
were obtained for each construct. Control transfectants
were generated using the empty CMV promoter construct.
Phosphorylation of STAT3 protein in BBCE cells
BBCE cells were seeded in 12-well plates (400 000 cells per
well), cultured for 2 days in DMEM containing 0.5% (v/v)
newborn calf serum and STAT3 and phospho-STAT3
(Tyr705) were detected as described in a kit purchased from
New England Biolabs.
Preparation of adenoviruses expressing STAT3 and
[
3
H]thymidine incorporation assays
Recombinant adenoviruses harbouring wild-type (AD/WT)
and dominant negative (Y705F) (AD/DN) Stat3 cDNAs as
well as a control adenovirus carrying only the vector (AD)
were prepared as described previously [26]. Adenoviruses
were amplified in 293 cells. The efficiency of infection and
the expression of the proteins were monitored by immuno-
fluorescence (Zeiss, Axiovert S100 microscope) and Western
blot analysis.
BBCE cells grown to subconfluency were infected with
recombinant adenoviruses at a multiplicity of infection of
5 : 1 for 2 h in full medium. The medium was renewed for
another 6 h allowing the constructs to be expressed and
then various concentrations of recombinant LIF (R & D
Systems) were added for an additional day. Then,
1lCiÆmL
)1
of [
3
H]methyl-thymidine (ICN) was added to
each of the wells for the last 4 h of the incubation. Culture
medium was removed and the cells were fixed with ice-cold
10% trichloroacetic acid for 20 min at 4 C, washed three
times with water and solubilized in 0.1
M
NaOH overnight
at 4 C. The radioactivity was counted in a beta liquid
scintillation counter (LKB).
RESULTS
SI.1 is identified as LIF
The flow-through of the initial cation exchange chroma-
tography step containing SI.1 activity [21] was sequentially
subjected to concanvalin A–Sepharose affinity chromatog-
raphy (Fig. 1A) and preparative SDS/PAGE tube electro-
phoresis (Fig. 1B). Following exchange to a nondenaturing
detergent, the activity was evaluated and the active fractions
were subjected to a final C
18
RP-HPLC chromatography
(Fig. 1C). The main SDS/PAGE band exhibited a large size
distribution (40–60 kDa) as a result of heterogeneity and
glycosylation (Fig. 1D). Sequence analysis of five different
segments of the main band revealed that one of the
candidate proteins was LIF, a cytokine that has been
previously shown to exhibit inhibitory activity on endothe-
lial cells (Table 1). The rest of the sequenced peptides
belonged to proteins that appeared irrelevant concerning
inhibition of endothelial cell proliferation (Table 1). Con-
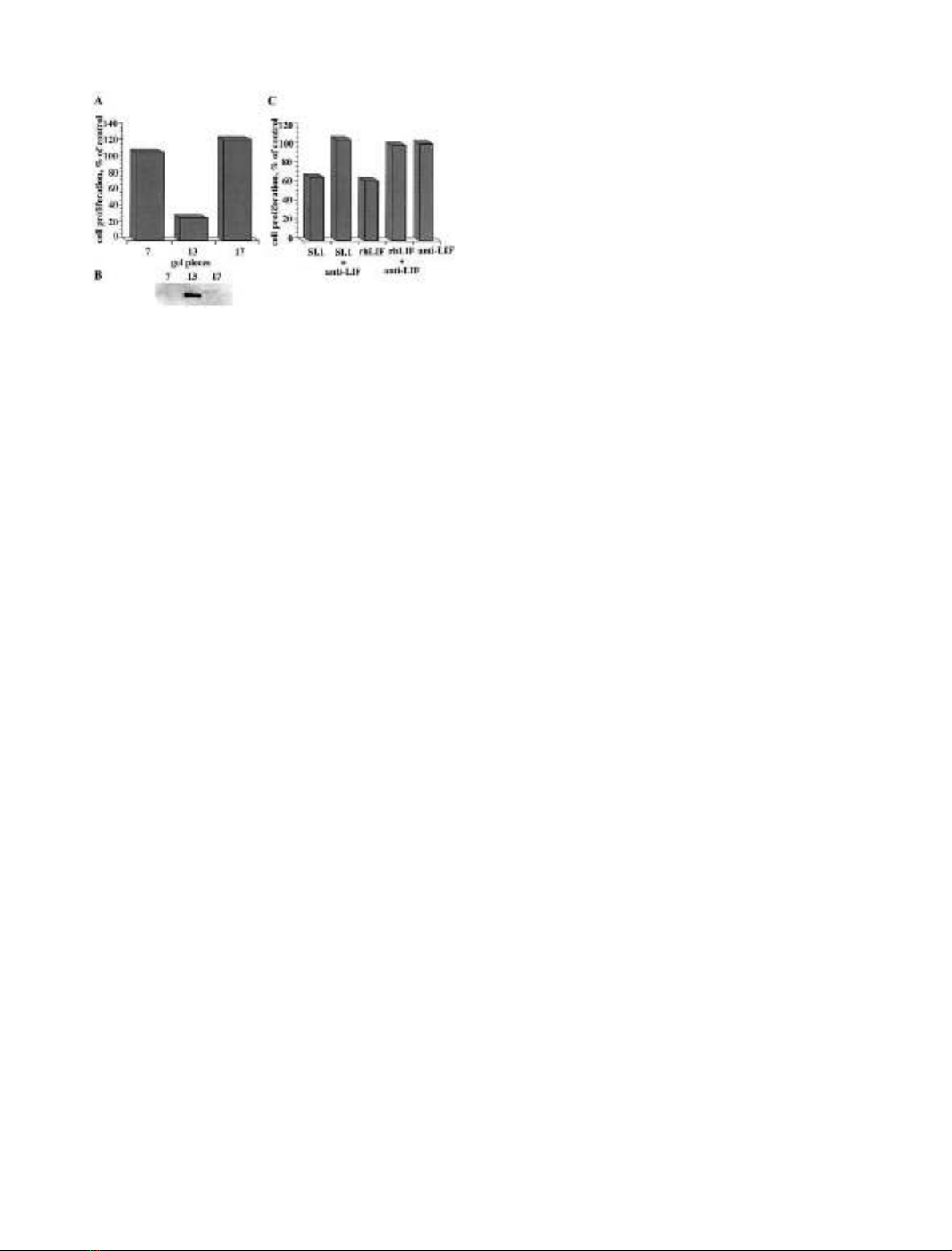
firmation of SI.1 identity as LIF was further obtained by
excising the corresponding SDS/PAGE gel segment, to that
containing the sequenced LIF peptides, from another lane
of the final SDS/PAGE gel. Following renaturation, this gel
segment showed both inhibitory activity on endothelial cells
(Fig. 2A) and the presence of LIF by Western blot analysis
with a specific LIF antibody (Fig. 2B). More importantly,
neutralizing LIF antibodies abolished the antiproliferative
SI.1 activity on endothelial cells (Fig. 2C).
LIF expression is down-regulated by N-Myc
Detection and isolation of LIF in SH-EP007 cells
(vs. WAC2) suggested that N-Myc down-regulated LIF.
To substantiate N-Myc-induced down-regulation of LIF,
we used promoter–reporter assays. Upon transfection of
SH-EP007 cells with a vector containing wild-type N-myc
cDNA (Fig. 3A), LIF promoter activity was suppressed
substantially (Fig. 3B). In order to identify the domain(s)
responsible for this inhibition, several deletions within the
N-myc cDNA were generated and tested (Fig. 3A). All
mutant N-Myc proteins were equally well expressed (data
not shown). Transfection of N-myc mutants lacking the
DNA-binding [d(381–395)] or the helix/loop
/
helix leucine
zipper [d(350–464)] regions inhibited LIF promoter-luc
activity, in a manner similar to the wild type N-myc.In
contrast, the N-terminal part of N-myc seemed to be
important for the inhibition of the LIF promoter transcrip-
tion as the mutants d(1–134) and d(1–300) had completely
lost this ability. As deletion of either the MbI [d(20–90)] or
the MbII [d(96–140)] domains, together with flanking
sequences, did not result in derepression of the inhibitory
activity (Fig. 3B), the results indicate that a contiguous
N-terminal N-Myc domain is essential for suppression of
the LIF promoter transcription. These data indicate an
N-myc-specific role in inhibiting LIF promoter activity.
LIF inhibits the proliferation of endothelial cells
but not that of WAC2 neuroblastoma cells
We had identified LIF due to its ability to inhibit an
important step of angiogenesis, i.e. vascular endothelial cell
proliferation. In agreement with this finding, in addition to
bovine brain capillary endothelial cells, recombinant human
LIF (rhLIF) strongly inhibited the bFGF-stimulated pro-
liferation of aorta (BAE) and human dermal microvascular
endothelial cells (Fig. 4A, and data not shown). However,
endothelial cells from other tissue origin, such as those from
adrenal cortex (ACE), were weakly inhibited (Fig. 4A). At
3734 E. Hatzi et al. (Eur. J. Biochem. 269)FEBS 2002
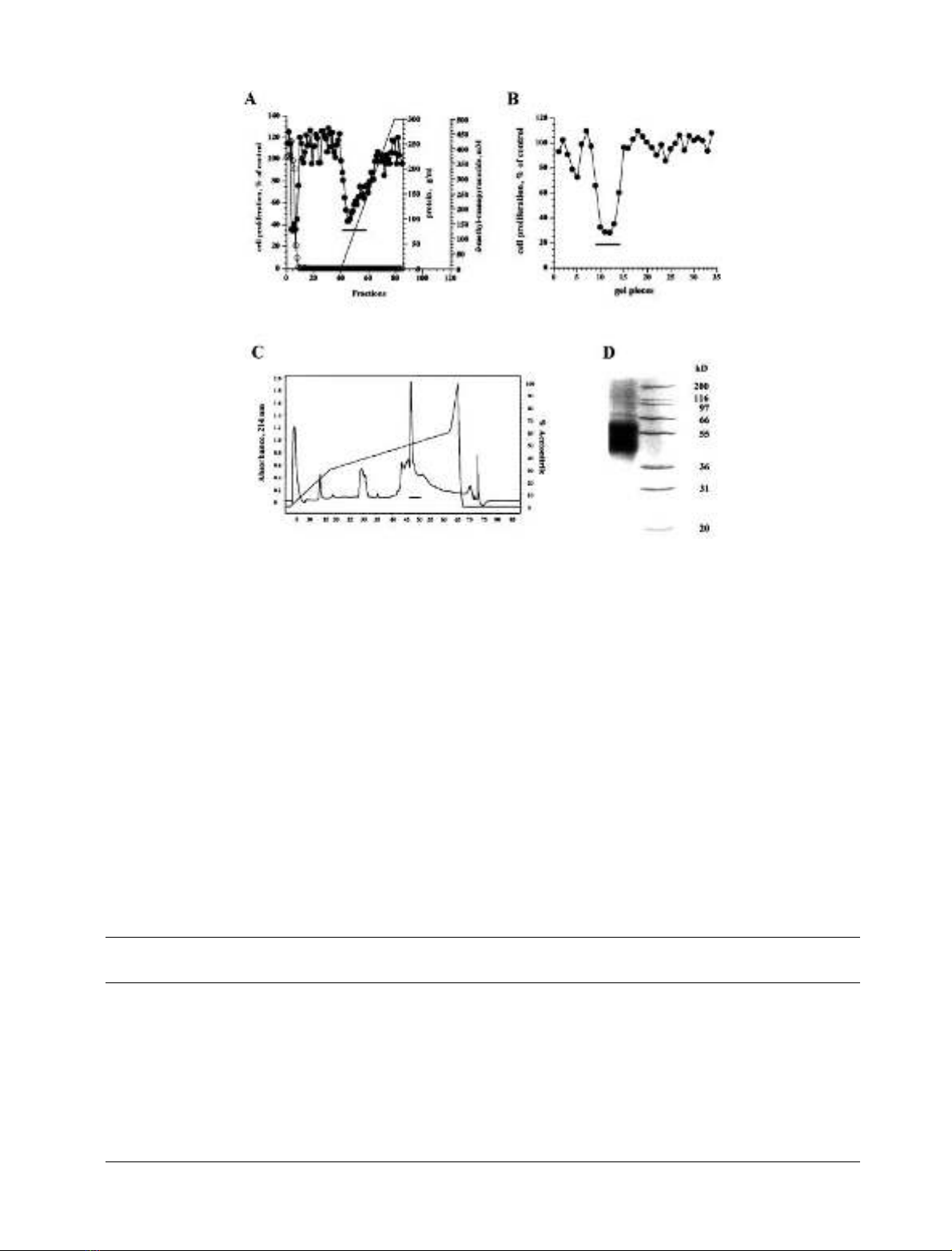
Fig. 1. Purification and isolation of SI.1. (A) The flow through fractions of the previous cation exchange chromatography step, containing SI.1, were
ultrafiltrated, lyophilized, dissolved in equilibration buffer and applied to a concanvalin A–Sepharose column. Bound material was eluted with
linear gradients of a-methyl-mannopyranoside as indicated (–). Aliquots of the fractions were tested for protein content (s) and activity on
endothelial (BBCE) cell proliferation (d). Ten microliters of the fractions were added every other day with 2.5 ngÆmL
)1
bFGF. The results were
expressed as percentage of control (cells receiving buffer only). (B) The active fractions from concanvalin A–Sepharose chromatography were
concentrated and diafiltrated using ultrafiltration and were subjected to SDS/PAGE in tubes as described under Material and methods. Following
electrophoresis, the tubes were washed with NaCl/P
i
/1% Chaps followed by a further 30-min wash in NaCl/P
i
. The 12% polyacrylamide tubes were
cut into 2.5-mm slices and a small piece of each slice was allowed to diffuse, overnight at 4 C with gentle agitation, in 700 lL of endothelial cell
medium. The diffusates were tested for inhibitory activity on endothelial cell proliferation (d). (C) The proteins of the active gel pieces were eluted in
dH
2
O and lyophilized for application onto a C
18
HPLC column. The application was performed in 0.1% TFA/dH
2
0 and elution was carried out
with a linear gradient from 0 to 30% acetonitrile in 10 min and 30–60% acetonitrile in 45 min. A flow rate of 1 mLÆmin
)1
was used and fractions of
1 mL were collected, lyophilized and resuspended into 100 lL culture-tested water for activity evaluation. Ten microliters of 100-fold dilutions of
the fractions were used for BBCE proliferation test (d). (D) The fractions corresponding to the active peak of the HPLC column in Fig. 1C were
pooled, lyophilized, and separated on a 12.5% SDS-polyacrylamide gel under reducing conditions. The bands were excised from Coomassie blue
stained gel and subjected to in-gel digestion followed by mass fingerprint analysis as described in material and methods. Left lane, pooled HPLC
fractions; right lane, molecular markers in kDa.
Table 1. Proteins identified by mass analysis and Edman sequencing of tryptic peptides. Thepositionofpeptideaminoacidinproteinwasdetermined
by Edman sequencing.
Band No.
number
Protein
position
SWISS-PROT
accession Amino-acid
1, minor Metabotropic glutamate
receptor 2
Q14416 20–34
Signal recognation particle P13624 29–43
2, major Alpha antitrypsin P34955 323–337
Carboxypeptidase H Prec P16870 87–107
3, major Carboxypeptidase H Prec P16870 148–157
Alpha antitrypsin P34955 205–213
4, major Leukemia inhibitory factor P15018 23–37, 61–80, 193–202, 202–107
5, minor Keratin I P13645
Keratin II P04264
FEBS 2002 N-myc oncogene down-regulates LIF (Eur. J. Biochem. 269) 3735
the concentrations used, rhLIF was not cytotoxic, as seen by
microscopic evaluation and by the fact that cell densities
never fell below those present at seeding. rhLIF also
inhibited basal proliferation of BBCE cells (data not shown
and Fig. 5). The inhibitory effect of LIF on endothelial cell
proliferation did not, however, exclude a direct autocrine
inhibitory effect of LIF on neuroblastoma cell proliferation.
Towards this end, rhLIF had no effect on the proliferation
of SH-EP007 and WAC2 neuroblastoma cells, even at very
high concentrations (Fig. 4B). The same results were
obtained when we transfected WAC2 neuroblastoma cells
with a vector containing the human LIF cDNA under the
control of a CMV promoter (not down-regulated by
N-Myc) compared to cells transfected with the empty
vector. The proliferation potential of the LIF-expressing
clones was either similar (c6) or higher (c11, c13) than the
control clones (cve-1 and cve-2) (Fig. 4C). Indeed, in the
case of c11 and c13 the proliferation rate was similar to that
of the parental WAC2 cells and clearly distinct from that of
the low N-myc expressing SH-EP007 cells (Fig. 4C). Thus,
forced overexpression of LIF in WAC2 cells did not inhibit
their in vitro proliferation.
LIF inhibits endothelial cell proliferation via the STAT3
pathway
LIF can mediate its effects by binding to specific cell surface
receptors with subsequent phosphorylation of the transcrip-
tion factor STAT3 at Tyr705. When rhLIF was added to
BBCE cells, it was, in fact, able to phosphorylate STAT3 in
a time-dependent manner (Fig. 5A). Phosphorylation of
STAT3 Tyr705 was observed at 2.5 min post induction,
reached a maximum by 20 min and declined slowly over
80 min. Simultaneous administration of angiogenic factors
such as bFGF did not alter the phosphorylation pattern of
STAT3 (data not shown).
We wished to determine whether the STAT3 pathway
was necessary and crucial for the LIF-induced inhibition of
vascular endothelial cell proliferation. To that aim, we
infected BBCE cells with recombinant adenoviruses con-
taining various stat3 forms and investigated the ability of
rhLIF to inhibit bFGF-induced proliferation of the infected
cells. rhLIF was able to inhibit proliferation of BBCE cells
infected with the empty vector controls (AD) or with the
vector containing the wild-type stat3 (AD/WT) (Fig. 5B). In
contrast, rhLIF was unable to inhibit proliferation of BBCE
cells infected with the dominant-negative stat3 mutant (AD/
DN) (Fig. 5B). Thus, the STAT3 pathway is important for
mediating the inhibitory signals of LIF regarding endothe-
lial cell proliferation.
DISCUSSION
We have previously shown that neuroblastoma cell super-
natants contained three endothelial cell proliferation inhib-
itors (SI.1, SI.2 and SI.3), the expression of which were
dramatically down-regulated upon N-Myc overexpression
[21]. In the present study, SI.1 activity was isolated from 47 L
of SH-EP007 supernatants by a series of chromatographic
steps using inhibition of endothelial cell proliferation as a
marker for biological activity. The main SDS/PAGE band
exhibited a large size distribution (40–60 kDa) as a result of
heterogeneity and glycosylation. Sequence analysis of
tryptic peptides combined with detection with specific
antibodies identified SI.1 as LIF. A conclusion further
supported by inhibition of the SI.1 activity by specific anti-
LIF neutralizing antibodies. Identification of SI.1 as LIF
strongly implied that N-Myc overexpression down-regulat-
ed LIF expression in neuroblastoma. Indeed, reporter assay
experiments revealed, for the first time, that N-Myc
overexpression dramatically down-regulated LIF promoter
transcription. LIF has never been reported to belong to a set
of genes shown to be regulated by the myc gene family
members using various assays [1,27], including crosslinking
[28], and cDNA microarrays [29].
Myc/Max interactions play an important role in Myc-
induced transcriptional regulation. Indeed, the C-terminus
of N-Myc contains the HLH and Zip domains, which
mediate heterodimerization with Max [2,30,31], and the BR
domain, which is required for binding of the N-Myc/
Max heterodimers to consensus sites known as E boxes
[30,32,33]. Our data suggest that down-regulation of LIF is
independent of N-Myc/Max interaction and DNA binding
as neither the HLH-Zip nor the BR deletion released the
transcriptional repression. This excludes an indirect repres-
sion of LIF gene via some of the known N-Myc/Max
regulated genes [1].
Our data, however, reserve an important role for the
N-terminal domain of N-Myc in down-regulation of LIF.
Indeed, deletion of amino acids 1–134 abolished almost all
of the N-Myc repressing activity. The N-terminal domain
contains Myc boxI (MbI) and Myc boxII (MbII), two
highly conserved domains of the myc family that are
considered important sites for protein interactions. In most
transcriptional assays, MbII domain is associated with
repression rather than transactivation [34,35]. Indeed, MbII
Fig. 2. Identification of SI.1 as LIF. A small fraction of the sample used
for the sequencing of SI.1 was subjected to the same SDS/PAGE
electrophoresis under nonreducing conditions and treated as in
Fig. 1B. The gel pieces were analyzed for cell growth inhibition and
correlation with LIF immunoreactivity. (A) Part of the eluates of the
gel pieces (presented in this figure the gel pieces 7, 13, 17) was exami-
nated for their ability to inhibit bFGF-stimulated proliferation of
BBCE cells. (B) Aliquots of the same eluates were analyzed by Western
blot using a goat anti-LIF Ig (1 lgÆmL
)1
). The bound antibodies were
detected with horseradish peroxidase conjugated anti-goat IgG fol-
lowed by ECL detection system. (C) Neutralization of SI.1 activity.
BBCE cells were treated either with rhLIF (2.5 ngÆmL
)1
) or aliquots of
the gel piece 13 alone or in combination with antibodies against LIF
(anti-LIF, 1 lgÆmL
)1
) as described in material and methods. The
results were expressed as percent of control (cells received only bFGF).
3736 E. Hatzi et al. (Eur. J. Biochem. 269)FEBS 2002


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)