
a-Fetoprotein positively regulates cytochrome
c
-mediated caspase
activation and apoptosome complex formation
Lidia Semenkova
1,
*, Elena Dudich
1,
*, Igor Dudich
1
, Natalie Tokhtamisheva
1
, Edward Tatulov
2
,
Yury Okruzhnov
3
, Jesus Garcia-Foncillas
3
, Juan-Antonio Palop-Cubillo
4
and Timo Korpela
5
1
Institute of Immunological Engineering, Moscow, Russia;
2
Anticancer Drug Research Center, Moscow, Russia; Departments of
3
Oncology and
4
Organic Chemistry and Pharmacology, University of Navarra, Pamplona, Spain;
5
Joint Finnish-Russian
Biotechnology Laboratory, Turku University, Finland
Previous results have shown that the oncoembryonic marker
a-fetoprotein (AFP) is able to induce apoptosis in tumor
cells through activation of caspase 3, bypassing Fas-
dependent and tumor necrosis factor receptor-dependent
signaling. In this study we further investigate the molecular
interactions involved in the AFP-mediated signaling of
apoptosis. We show that AFP treatment of tumor cells is
accompanied by cytosolic translocation of mitochondrial
cytochrome c. In a cell-free system, AFP mediates process-
ing and activation of caspases 3 and 9 by synergistic
enhancement of the low-dose cytochrome c-mediated sig-
nals. AFP was unable to regulate activity of caspase 3 in cell
extracts depleted of cytochrome cor caspase 9. Using
high-resolution chromatography, we show that AFP posit-
ively regulates cytochrome c/dATP-mediated apoptosome
complex formation, enhances recruitment of caspases and
Apaf-1 into the complex, and stimulates release of the active
caspases 3 and 9 from the apoptosome. By using a direct
protein–protein interaction assay, we show that pure human
AFP almost completely disrupts the association between
processed caspases 3 and 9 and the cellular inhibitor of
apoptosis protein (cIAP-2), demonstrating its release from
the complex. Our data suggest that AFP may regulate cell
death by displacing cIAP-2 from the apoptosome, resulting
in promotion of caspase 3 activation and its release from the
complex.
Keywords: apoptosis; apoptosome; cytochrome c;IAP-2;
a-fetoprotein.
Apoptotic cell death is characterized by biochemical and
morphological changes, which are largely caused by caspase
activity. A class of cysteine proteases, known as caspases,
which are constitutively expressed in cells as inactive
proenzymes, require proteolytic cleavage to be activated.
In general, either receptor-induced or mitochondrion-
induced death signals stimulate activation of specific
adapterproteinsFADD/MORT1orApaf-1byformation
of the high-molecular-mass death-inducing complex or
apoptosome. The adapter proteins recruit initiator caspases
8 and 9 to activate them by autoprocessing. Once activated,
initiator caspases are ready to induce processing of down-
stream effector caspases 3 and 7 [1]. The mitochondrial
apoptosis pathway is mediated by cytochrome c(cyt-c)
release with the subsequent formation of the Apaf-1/cyt-c/
dATP/procaspase 9 apoptosome complex, leading to acti-
vation of caspase 9 and downstream effector caspases [2].
Chromatographic analysis of the apoptosome assembly
indicated that, in native cell lysates, Apaf-1 oligomerizes
into multimeric complexes of molecular mass 1.4 MDa
and 700 kDa, which in addition to processed caspase 9,
contain fully processed caspase 3 and 7 [3]. Caspases are
inhibited by a number of cellular inhibitor of apoptosis
proteins (cIAPs), which bind directly to procaspases 9 and 3
to prevent their cyt-c-mediated processing and activation
[4,5]. During apoptosis, a mitochondrial protein named
Smac/DIABLO [6] that directly binds to IAPs to remove
them from the apoptosome complex [4,7], cancels the
IAP-mediated caspase inhibition. Recently, another IAP-
inhibitory protein Omi/HtrA2 was characterized, which
operates by abrogation of the IAP–caspase interaction [8].
AFP is the major serum protein of embryonic plasma
that is involved in regulation of gene expression, differen-
tiation, proliferation and apoptosis in developing cells
[9–12]. Although, the biological role of this protein is not
yet fully understood, it has been well characterized as a
physiological carrier/transport protein for various ligands,
including fatty acids, drugs, hormones, heavy metals,
delivering them to developing and malignant cells [9,12].
The specific expression and internalization of AFP is
restricted to developing cells, such as embryonic cells,
activated immune cells and tumor cells, which suggests its
important regulatory role in cell growth and differentiation
[9,10,12]. Various researchers have documented the exist-
ence of specific receptor-dependent mechanisms responsible
for the active endocytosis of AFP by malignant cells [13,14].
Microscopic data have demonstrated that fluoresceinated
Correspondence to E. Dudich, Institute of Immunological Engineering,
142380, Lyubuchany, Moscow Region, Chekhov District, Russia.
Tel./Fax: + 7 095 996 15 55, E-mail: dudich@ineos.ac.ru
Abbreviations: AFP, a-fetoprotein; cyt-c, cytochrome c; cIAP, cellular
inhibitor of apoptosis protein; Ac-DEVD-AMC, Ac-Asp-Glu-Val-
Asp-7-amino-4-methylcoumarin; LEHD-AFC, Leu-Glu-His-Asp-
aminotrifluoromethylcoumarin; IETD-AMC, Ile-Glu-Thr-Asp-7-
amino-4-methylcoumarin; CHO, aldehyde.
*Note: These authors contributed equally to this work.
(Received 11 February 2003, revised 28 August 2003,
accepted 16 September 2003)
Eur. J. Biochem. 270, 4388–4399 (2003) FEBS 2003 doi:10.1046/j.1432-1033.2003.03836.x

AFP is specifically bound to the cell surface at 4 Cand
internalized into the cytoplasm at 37 C [15,16]. It has been
shown that AFP is internalized via coated pits and vesicles
before being delivered to endosomes [15,16]. Much evidence
of cell growth regulatory activity, including tumor suppres-
sion, has been reported for various species of the full-length
AFP molecule [17–22], its proteolytic fragments [23],
recombinant domains [24] and synthetic peptides [25–27].
It has been demonstrated that AFP realizes its tumor-
suppressive activity by triggering apoptosis, characterized
by typical morphological changes, growth arrest, cytotoxi-
city, and DNA fragmentation [20–22]. It was shown that
AFP induces apoptosis in malignant cells through activa-
tion of caspase 3, bypassing Fas/FasL and tumor necrosis
factor (TNF)/TNFR-dependent pathways and does not
require upstream activation of receptor-dependent initiatory
caspase 8 and caspase 1 [21]. Although these studies have
shown that a caspase cascade is initiated during AFP-
induced apoptosis, the mechanisms by which AFP triggers
caspase activation are unknown. Our previous experimental
data show that AFP does not require de novo protein
synthesis and RNA expression to trigger apoptosis, as it was
not blocked by actinomycin D or cycloheximide [20].
In this study, we aimed to determine how AFP activates
the caspase cascade. To understand the molecular mecha-
nisms of AFP-mediated apoptosis signaling, we established
a cell-free system, similar to that used for studies of cyt-c-
induced apoptosis [28,29]. We show here that AFP syner-
gistically enhances caspase activation and processing in the
presence of a low suboptimal dose of cyt-c and requires the
presence of all members of the apoptosome complex to
initiate this process. We examine the mechanisms by which
AFP regulates apoptosis and demonstrate that the pro-
apoptotic effect of AFP is mediated through its interaction
with apoptosome-forming proteins. Chromatographic ana-
lysis of the apoptosome assembly demonstrated that AFP
stimulates formation of the Apaf-1–apoptosome complex,
enhances recruitment and activation of procaspase 3 in the
complex, and stimulates release of active caspase 3 and 9
from the apoptosome. Our data suggest that AFP may
regulate cell death by displacing cIAP-2 from the apopto-
some complex, thereby promoting caspase 3 release from
the complex.
Materials and methods
AFP purification
Human AFP was isolated from the cord serum using ion-
exchange, affinity and gel-filtration chromatography as
described previously [23]. AFP purity was established using
PAGE and immunoblotting with monospecific antibodies
against human AFP and adult serum proteins and was
showntobenolessthan99.8%.
Cells
HepG2 cells originated from the American Type Culture
Collection were cultured in Dulbecco modified Eagle’s
medium (ICN Biomedicals) with
L
-glutamine and 10%
heat-inactivated fetal bovine serum, 100 IU penicillinÆmL
)1
,
0.1 mg streptomycinÆmL
)1
in a humidified 5% (v/v)
atmosphere of CO
2
at 37 C. For a passage, cells were
incubated in 0.25% (v/v) trypsin solution, then washed and
plated out.
Cytotoxicity assay
HepG2 cells were incubated with 5–7 l
M
AFP for deter-
mined time intervals of 2–14 h, and then assessed for their
viability by the trypan blue exclusion assay as described
previously [22]. Cells cultivated without additions were
taken as a control. The experimental data were expressed as
the percentage of dead cells relative to the total amount of
cells.
Preparation of cell-free extracts
Cell-free S-100 extracts were generated from human
hepatocarcinoma HepG2 as described [29,30]. Cells
(4 ·10
8
) were collected and washed (three times) in
50 mL NaCl/P
i
and once in 5 mL hypotonic cell extraction
buffer (containing 20 m
M
Hepes, pH 7.2, 10 m
M
KCl,
2m
M
MgCl
2
,1m
M
dithiothreitol, 5 m
M
EGTA, 25
lgÆmL
)1
leupeptin, 5 lgÆmL
)1
pepstatin, 40 m
M
b-glycero-
phosphate, 1 m
M
phenylmethanesulfonyl fluoride). The cell
pellet was then resuspended in an equal volume of cell
extraction buffer, allowed to swell for 20 min on ice, and
then disrupted by 30–50 strokes of a Dounce homogenizer.
The homogenate was centrifuged at 3000 gfor 10 min at
4C to remove whole cells and nuclei. The supernatant was
centrifuged at 15 000 gfor 20 min at 4 Candthen,to
obtain the cytosolic S-100 extract, the supernatant was
re-centrifuged at 100 000 gfor 1 h at 4 C. Extracts were
assessed for protein content by the Bradford assay and
stored in aliquots at )70 C. Cyt c-free cytosolic extracts
were prepared in more mild conditions by the slightly
modified procedure described in [30].
In vitro
caspase activation
For in vitro caspase activation, 40 lg of the S-100 extract
(complete or after immunodepletion) was incubated for the
indicated times with bovine heart cyt-c (Sigma-Aldrich,
St Louis, MO, USA) and/or pure human AFP (5 l
M
)inthe
presence or absence of 1 m
M
dATP (Sigma) in 15 lLofa
reaction buffer (10 m
M
Hepes, pH 7.2, 25 m
M
NaCl, 2 m
M
MgCl
2
,5m
M
dithiothreitol, 5 m
M
EDTA, 0.1 m
M
phenyl-
methanesulfonyl fluoride) at 30 C. To control specificity
of AFP effects, the equivalent amount of human serum
albumin (Sigma) was added instead of AFP. The activity and
proteolytic processing of caspases 3 and 9 were then detected
by fluorimetric assay and immunoblotting with the corres-
ponding antibodies supplied by Santa Cruz Biotechnology,
Inc (Santa Cruz, CA, USA): polyclonal goat anti-(caspase 3)
p20 (N19); anti-(caspase 3) p11 (K19); rabbit anti-
(caspase 9) p10 (H-83); rabbit anti-(caspase 9) p35 (H-170).
Fluorimetric assay of caspase activity
Caspase activities were determined by incubation of the
extract aliquots (5 lL) for various times at 30 C with one
of the fluorogenic substrates [40 l
M
Ac-DEVD-AMC
(ICN Biomedicals Inc), 50 l
M
LEHD-AFC (Chemicon
FEBS 2003 AFP amplifies cytochrome c-mediated caspase activation (Eur. J. Biochem. 270) 4389

International, Temecula, CA, USA) or 50 l
M
IETD-AMC
(Alexis Biochemicals, San Diego, USA] in 16 lL substrate
buffer (25 m
M
Hepes, pH 7.2, 100 m
M
NaCl, 1 m
M
EDTA,
0.1% Chaps, 10 m
M
dithiothreitol, 10% sucrose). Reactions
were terminated by dilution with 2.0 mL ice-cold 0.2 m
M
sodium phosphate buffer, pH 7.5, and fluorescence was
measured using a Perkin–Elmer MPF-44A fluorimeter
(k
exc
¼365 nm and k
em
¼440 nm for the AMC fluores-
cence or k
exc
¼400 nm and k
em
¼505 nm for the AFC
fluorescence). For each sample, caspase activity was
expressed in relative units, pmolÆmin
)1
Æmg
)1
, showing the
amount of cleaved substrate in pmol normalized for time of
reaction with substrates and cytosolic protein concentra-
tion, or in relative fluorescent units (FU) per fraction.
Immunoprecipitation and immunoblotting analysis
S-100 cytosolic extracts obtained from HepG2 cells were
immunodepleted from endogenous cyt-c, procaspase 9 or
procaspase 3 by immunoprecipitation with the corres-
ponding antibodies as described [31]. Briefly, 50 lLofthe
S-100 cell extract (4–5 mgÆmL
)1
; reaction buffer with
addition of 0.1% Chaps) was incubated for 2 h at 4 C
with 5 lg of the corresponding antibodies: anti-cyt-c
6H2.B4 (PharMingen, San Diego, CA, USA), anti-
(caspase 9) clones C-18 and H-83 or anti-(caspase 3)
(N-19). The control cell extracts were incubated with the
equivalent amounts of the control antibodies of the same
type. Immune complexes were precipitated by addition of
antibody/extract mixture on to drained protein G-Seph-
arose or protein A/agarose beads (Amersham Pharmacia
Biotech) for 2 h at 4 C. Coated beads were then
removed by centrifugation, and the resulting immuno-
depleted lysates after adjustment for protein concentration
were used immediately for caspase activation experiments.
The extent of depletion was controlled by immunoblot-
ting with the corresponding antibodies. Immunoblotting
with b-actin antibodies (ICN Biomedicals Inc) was
performed as a loading control.
For immunoblotting analysis, protein samples (50 lgper
lane) were subjected to standard SDS/PAGE in a 12% or
15% polyacrylamide gel and transferred on to 0.45-l
M
poly(vinylidene difluoride) membranes by semidry electro-
blotting, followed by probing for various proteins using the
corresponding antibodies: rabbit anti-(Apaf-1), H-324
(Santa Cruz); affinity-purified rabbit anti-(human cIAP-2),
HIAP-1 (R & D Systems, Wiesbaden, Germany); rabbit
polyclonal anti-(caspase 8) p20, H-134 (Santa Cruz) or the
corresponding polyclonal antibody goat anti-(caspase 3) or
anti-(caspase 9). Bound antibodies were detected using
appropriate horseradish peroxidase-conjugated anti-rabbit
or anti-goat secondary IgGs (Santa Cruz) and developed by
enhanced chemiluminescence staining using ECL reagents
(Amersham Pharmacia Biotech). Gel calibration was per-
formed with the Low Molecular Weight Calibration Kit for
SDS Electrophoresis (Amersham Pharmacia Biotech).
Dot-blot analysis was performed as usual. Briefly, 1-lL
aliquots taken from the chromatographic fractions were
applied to the nitrocellulose membranes, then blocked by
defatted milk. The membranes were then probed with rabbit
polyclonal affinity-purified anti-(human AFP) IgG. Bound
antibodies were detected using appropriate peroxidase-
coupled secondary antibodies and developed as described
above.
Assay of cyt-c release
Cyt-c translocation from mitochondria to the cytoplasm
was assessed by direct immunochemical measurement of the
cyt-c in the cytosolic and mitochondrial fractions obtained
from HepG2 cells treated with AFP for various time
intervals. Briefly, cells (0.5 ·10
6
cells per well) in Dulbecco’s
modified Eagle’s medium with 10% fetal bovine serum were
plated on the flat-bottomed 24-well plates (Nunc) and
incubated for 24 h. Then 5 l
M
AFP was added to each well.
After various lengths of treatment (2–17 h), cells were
scraped, washed in NaCl/P
i
, and resuspended in 200 lL
digitonin lysis buffer (0.025% digitonin in 250 m
M
sucrose,
20 m
M
Hepes, pH 7.4, 5 m
M
MgCl
2
,10m
M
KCl, 1 m
M
EDTA, 1 m
M
EGTA, 10 m
M
Tris/HCl, pH 7.4,
10 lgÆmL
)1
leupeptin, 10 lgÆmL
)1
aprotinin, and 1 m
M
phenylmethanesulfonyl fluoride) [32]. After 10 min, cell
lysates were centrifuged for 2 min at 14 000 gat 4 Cto
obtain the supernatant (cytosolic fraction) and the pellet
(mitochondrial fraction). Mitochondrial pellet was solubi-
lized by a 30-min incubation with 100 lL lysing buffer
(150 m
M
NaCl, 1% Nonidet P40, 0.5% deoxycholate, 0.1%
SDS, 50 m
M
Tris/HCl, pH 7.5, cocktail of protease inhi-
bitors). Thereafter, cellular debris was removed by a 10-min
centrifugation at 14 000 gat 4 C. The supernatant com-
prising the membrane fraction was retained. Equal amounts
of cytosolic extracts and solubilized mitochondrial pellets
(50 lg protein) were fractionated by SDS/PAGE using 15%
polyacrylamide and then analysed by Western blot using the
cyt-c antibody 7H8.2C12, cyt-c oxidase subunit II antibody
(Molecular Probes), and b-actin antibody and ECL as
described above.
Direct protein–protein interaction assay
To determine possible interactions between AFP and
caspase 3, caspase 9 and cIAP-2, we used a direct copre-
cipitation assay with purified proteins. Before the experi-
ments, 25 lL Ni/Sepharose beads (Qiagen, Valencia, CA,
USA) were incubated for 1 h at 20 C in a solution of assay
buffer (50 m
M
Tris/HCl, 100 m
M
KCl, 10% sucrose, 0.1%
Chaps, 0.5 m
M
dithiothreitol, pH 7.4), containing 1%
ovalbumin, 12 lg His-tagged human recombinant
caspase 9 and 3 lg active His-tagged rat recombinant
caspase 3 (Alexis Biochemicals). After being washed, one
half of the beads was added to the cytosolic extract of
HepG2 cells (500 lg total protein) together with 20 lgAFP
andincubatedfor2hat4C. The control beads were
incubated with the same amount of HepG2 cytosolic extract
without AFP addition. The protein–bead complexes were
then washed (four times), isolated by centrifugation, boiled
in 15 lL sample buffer, and analyzed by SDS/PAGE/
Western blotting with anti-cIAP2 (HIAP-1) IgG.
Chromatographic analysis of the apoptosome assembly
To study effects of AFP on recruitment, processing and
release of various caspases from apoptosome and micro-
apoptosome complexes, we used the previously described
4390 L. Semenkova et al.(Eur. J. Biochem. 270)FEBS 2003

gel filtration technique [3]. Briefly, S-100 extracts were
prepared from HepG2 cells (6 mgÆmL
)1
) and activated by a
1-h incubation at 30 C with 1.0 m
M
dATP/1.5 m
M
MgCl
2
/
1.0 l
M
cyt-c with or without 5.0 l
M
AFP. Before addition
to the S-100 extracts, AFP samples were dialyzed against the
elution buffer. Activated lysate proteins (1mg) were
applied (0.2 mLÆmin
)1
;4C) to a 10/30 Superose-6 HR
column connected to an FPLC system (Amersham Phar-
macia Biotech). The column was eluted with elution buffer
(20 m
M
Hepes/KOH, 10 m
M
KCL, 1 m
M
EDTA, 1 m
M
EGTA, 1 m
M
dithiothreitol, 1.5 m
M
MgCl
2
,0.01m
M
phenylmethanesulfonyl fluoride, pH 7.2); 1-mL fractions
were collected. Aliquots of the fractions were taken for
measurement of caspase activity using the corresponding
fluorogenic substrates: DEVD-AMC for caspase 3 and Ac-
IETD-AMC for caspases 9 and 8 [33] as described above.
Fractions were then concentrated 20-fold with 2 mL
centrifugal concentrators (Centricon YM-10; Amicon) and
analyzed by PAGE and immunoblotting for changes in
distribution of AFP, Apaf-1, cIAP-2, caspases 3, 9 and 8.
Column calibration was performed with Gel Filtration
LMW and HMW calibration kits (Amersham Pharmacia
Biotech).
Results
AFP induces release of mitochondrial cyt-c in HepG2 cells
Our previous publications were devoted to the study of
AFP-induced apoptosis in whole cells and suggested that
this mechanism is independent of membrane receptor
signaling [20–23]. We investigate here the intracellular
molecular pathways of the AFP-mediated triggering of
apoptosis. To analyse the involvement of cyt-c release in
AFP-mediated apoptosis, cytosolic and mitochondrial
fractions were obtained from AFP-treated HepG2 cells
and analysed by Western blot for the presence of cyt-c. As
shown in Fig. 1, AFP induced the appearance of cyt-c in the
cytosolic fraction of treated HepG2 cells and its disappear-
ance from the mitochondrial fraction of treated cells,
indicating that AFP induced mitochondrial cyt-c release.
These data do not show, however, whether AFP induces
cytosolic cyt-c release directly or by indirect mechanisms by
activation of unknown factors.
AFP synergistically enhances low-dose cyt-c-mediated
caspase activation in cell-free cytosolic extracts
The mitochondrial apoptotic pathway could be activated by
addition of dATP to cell extracts to initiate the Apaf-1/
procaspase 9/cyt-c apoptosome cascade [28]. To determine
whether AFP is involved in this process, we established a
typical cell-free system using HepG2 cells and measured
caspase activation in this system with or without addition
of AFP. Two types of cell lysate were used for these
experiments: a typical S-100 cytosolic extract and a cyt-c-
free cytosolic extract, prepared by a mild procedure as
described previously [30]. Addition of AFP to the S-100
cytosolic extract triggered dATP-dependent induction of
caspase 3-specific DEVDase activity, which progressively
increased for at least 2 h (Fig. 2A). As a control, the
equivalent amount of human serum albumin was added to
the same cell-free system. No effect was observed at the level
of DEVDase activity. A low level of DEVDase activity was
also induced by dATP alone, evidently due to the presence
of a small amount of endogenous cyt-c in the preparations.
In the absence of dATP, AFP did not induce any caspase 3-
specific DEVDase activity at all.
To determine whether AFP can directly induce caspase
activation in cell-free cytosolic extract or requires the
presence of the basal level of cyt-c, we examined DEVDase
cleavage activity after addition of exogenous cyt-c and AFP
to the silentcytosolic extracts with undetectable endo-
genous cyt-c. Figure 2B shows that no DEVDase activity
was detected in this type of cytosolic lysate stimulated with
dATP/AFP or with dATP and low suboptimal dose of cyt-c
even 1.5 h after treatment. A significant time-dependent
increase in DEVDase activity was observed in the same
reaction system only after addition of all three compounds:
AFP,dATPandcyt-c(Fig.2B).ThelowDEVDaseactivity
in this experimental system compared with that described in
Fig. 2A is explained by the negligible amount of cyt-c in the
cytosol. These data demonstrate the ability of AFP to
amplify caspase-activating signals induced by low subopti-
mal doses of cyt-c.
We then examined the effect of AFP on the DEVDase
activity mediated by different doses of cyt-c in S-100
extracts. Figure 2C shows that, similarly to the above data
(Fig. 2A), AFP synergistically enhances DEVDase activity
induced by low suboptimal doses of cyt-c. A further increase
in cyt-c concentration in the cell extract resulted in the
saturationeffect, when the maximal stimulation of
caspase 3-specific DEVDase activity was reached, which
AFP cannot further increase (Fig. 2C).
Fig. 1. Effect of AFP on cell viability and cyt-c release in HepG2 cells.
HepG2 cells were treated with 5 l
M
AFP for various time intervals,
and then cytosolic and mitochondrial extracts were prepared at the
indicated times. Equal amounts of cytosolic and mitochondrial
extracts (50 lg) were immunoblotted with anti-(cyt-c) to assess cyt-c
release. b-Actin and cytochrome oxidase subunit II (Cyt ox.) were also
analysed in cytosolic and mitochondrial extracts as controls for protein
loading. Cell viability of AFP-treated HepG2 cells was assessed by the
trypan blue exclusion assay as described in Materials and methods.
FEBS 2003 AFP amplifies cytochrome c-mediated caspase activation (Eur. J. Biochem. 270) 4391
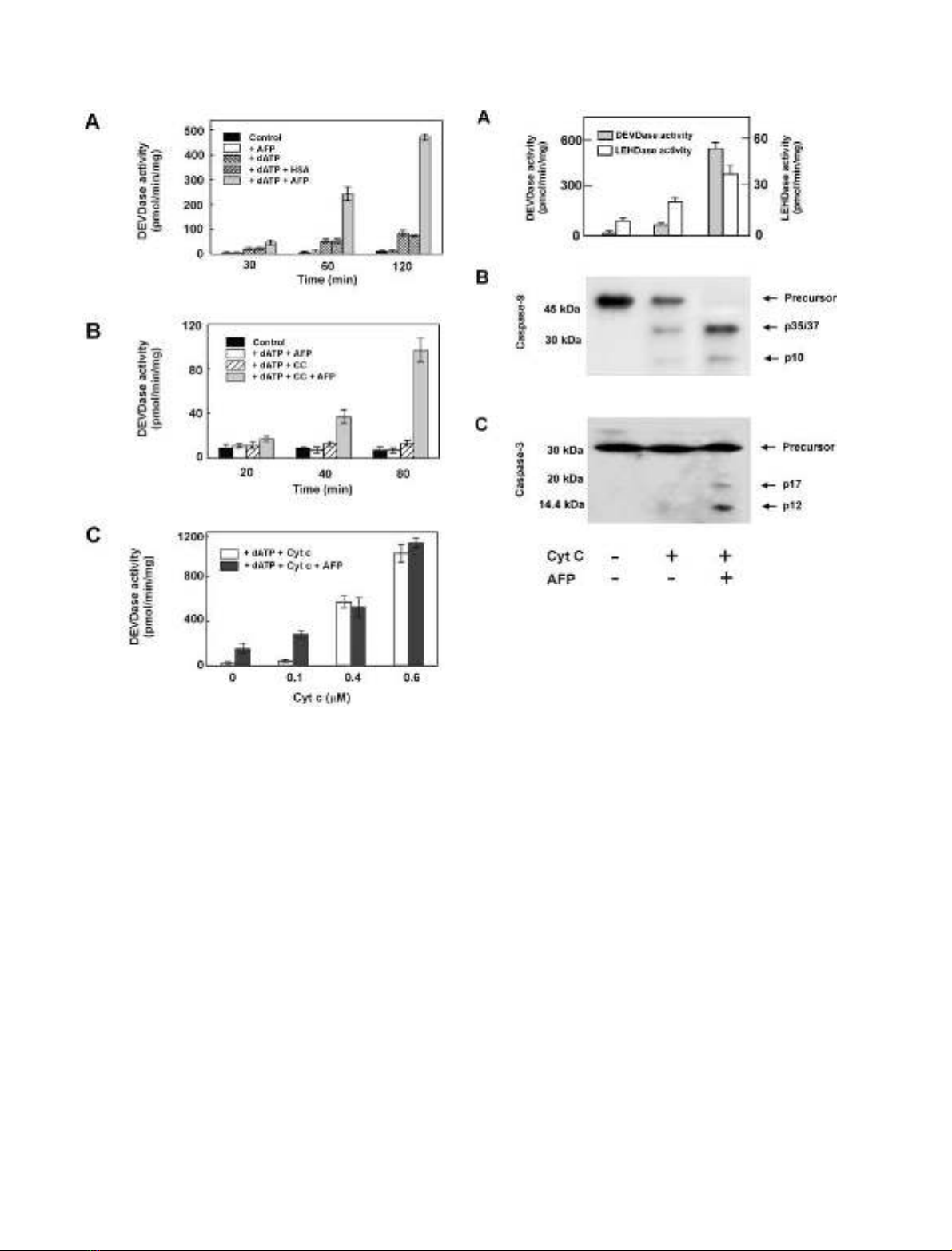
AFP synergistically enhances cyt-c-mediated processing
and activation of procaspases 9 and 3 in cell-free
cytosolic extracts
To determine whether AFP could induce increased caspase
activation in a cell-free system, we examined S-100 extracts
for cleavage of procaspases 3 and 9 and corresponding
fluorogenic caspase substrates after addition of AFP/cyt-c/
dATP. Both procaspase 9 and procaspase 3 were processed
to their active forms, giving the corresponding fragments
p35/37 and p10 for caspase 9 and p17 and p12 for
caspase 3. However, when AFP was combined with cyt-c/
dATP, more complete cleavage of the procaspases was
observed (Fig. 3B,C). In addition, there was a dramatic
increase in caspase 3-like DEVDase activity and a notable
increase in caspase 9-like LEHDase activity on combined
treatment with AFP/cyt-c/dATP in comparison with cyt-c/
dATP (Fig. 3A). These data show that AFP positively
regulates both processing and activation of procaspases 9
and 3 in cell-free cytosolic extracts by amplification of the
low-dose cyt-c-mediated effects.
AFP induces caspase activation only in the presence
of the all components of the apoptosome complex
The above experiments demonstrated functional interfer-
ence of AFP with the cyt-c-mediated process of caspase
activation. We studied further the functional significance of
Fig. 2. AFP enhances cyt-c-mediated DEVDase activity in cell-free
cytosolic extracts. (A) AFP induces caspase 3 activation in cell-free
S-100 cytosolic extracts in the presence of dATP. Effect of endogenous
cyt-c. Aliquots of HepG2-derived cytosolic extract (25 lgprotein)
were treated for various times with AFP (5 l
M
) or as a control with the
same dose of human serum albumin in the presence of dATP (1 m
M
)
and then assayed for DEVDase activity. (B) Synergistic increase in
DEVDase activity mediated by AFP in cyt-c-free cytosolic extracts on
addition of exogenous cyt-c. Aliquots of the cyt-c-free HepG2-derived
cytosolic extracts (25 lg protein) were treated for various times with
AFP (5 l
M
), cyt-c (0.2 l
M
) or a combination of the same doses of the
two compounds in the presence of dATP (1 m
M
) and then assayed for
DEVDase activity. (C) AFP differently affects caspase 3 activation in
cell-free cytosolic extracts induced by various doses of cyt-c. Aliquots
of S-100 cytosolic extract (25 lg protein) were treated for 30 min with
AFP (5 l
M
) and various doses of cyt-c in the presence of dATP (1 m
M
)
and then assayed for DEVDase activity. The mean ± SD from four
determinations is shown.
Fig. 3. AFP positively regulates cyt-c-mediated DEVDase and LEH-
Dase activity and processing of procaspase 9 and 3 in a cell-free system.
Aliquots of HepG2-derived S-100 cytosolic extract with addition of
1m
M
dATP were treated in the presence (+) or absence (–) of cyt-c
(0.2 l
M
) and/or AFP (5 l
M
). (A) Proteolytic activities of caspase 9 and
3 in experimental lysates were assayed by monitoring the cleavage of
the corresponding fluorogenic substrates LEHD-AFC and Ac-DEVD-
AMC. The mean ± SD from four determinations is shown.
Processing of caspases was detected by immunoblotting with the cor-
responding antibodies that recognize the precursors and subunits of
active caspase 9 (B) and 3 (C).
4392 L. Semenkova et al.(Eur. J. Biochem. 270)FEBS 2003


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)