
The fate of newly synthesized V-ATPase accessory subunit Ac45
in the secretory pathway
Vincent Th. G. Schoonderwoert, Eric J. R. Jansen and Gerard J. M. Martens
Department of Animal Physiology, Nijmegen Center for Molecular Life Sciences, University of Nijmegen, the Netherlands
The vacuolar H
+
-ATPase (V-ATPase) is a multimeric
enzyme complex that acidifies organelles of the vacuolar
system in eukaryotic cells. Proteins that interact with the
V-ATPase may play an important role in controlling the
intracellular localization and activity of the proton pump.
The neuroendocrine-enriched V-ATPase accessory subunit
Ac45 may represent such a protein as it has been shown to
interact with the membrane sector of the V-ATPase in only a
subset of organelles. Here, we examined the fate of newly
synthesized Ac45 in the secretory pathway of a neuroendo-
crine cell. A major portion of intact 46-kDa Ac45 was
found to be N-linked glycosylated to 62 kDa and a minor
fraction to 64 kDa. Trimming of the N-linked glycans
gave rise to glycosylated Ac45-forms of 61 and 63 kDa
that are cleaved to a C-terminal fragment of 42–44 kDa (the
deglycosylated form is 23 kDa), and a previously not
detected 22-kDa N-terminal cleavage fragment (the
deglycosylated form is 20 kDa). Degradation of the
N-terminal fragment is rapid, does not occur in lysosomes
and is inhibited by brefeldin A. Both the N- and C-terminal
fragment pass the medial Golgi, as they become partially
endoglycosidase H resistant. The Ac45 cleavage event is a
relatively slow process (half-life of intact Ac45 is 4–6 h) and
takes place in the early secretory pathway, as it is not affected
by brefeldin A and monensin. Tunicamycin inhibited
N-linked glycosylation of Ac45 and interfered with the
cleavage process, suggesting that Ac45 needs proper folding
for the cleavage to occur. Together, our results indicate that
Ac45 folding and cleavage occur slowly and early in the
secretory pathway, and that the cleavage event may be linked
to V-ATPase activation.
Keywords: acidification; regulated secretory pathway; post-
translational modification; vacuolar proton ATPase;
Xenopus.
Acidification of organelles in eukaryotic cells is required for
a variety of cellular processes, such as the release of ligands
from receptors during endocytosis and the hydrolysis of
macromolecules in lysosomes [1–3]. In the secretory path-
way, the lumen gradually acidifies from endoplasmic
reticulum (ER) to Golgi to secretory granules (reviewed in
[4]). The pH of the lumen of the ER, Golgi, and trans-Golgi
Network (TGN) is 7.3, 6.4, and 6.0, respectively, and
is similar in regulated and nonregulated secretory cells
[5–10]. The significance of the pH in the ER remains to be
established, although it seems likely that ER processes such
as protein glycosylation and folding depend on it. The low
pH in the Golgi has been shown to be important for the
regulation of protein–protein interactions [11,12] and the
activity of the N-glycan processing enzyme sialyltransferase
[13]. In the TGN, an acidic pH is necessary for the proper
processing of proproteins [14] and for the condensation of
regulated secretory proteins, which is important for their
targeting to immature secretory granules [15–17]. Immature
secretory granules mature and become progressively more
acidic (pH of 5.5 [18–20]). Granular acidification further
concentrates regulated proteins [21], while nonregulated
proteins are sorted away into clathrin-coated vesicles that
pinch off from the maturing granule [22–24]. Furthermore,
the acidic granular pH is necessary for the processing
enzymes to efficiently cleave the prohormones [25].
Acidification of intracellular compartments is established
and maintained by the vacuolar H
+
-ATPase (V-ATPase).
This multimeric enzyme complex consists of at least 13
different subunits that have been classified into a membrane
integral sector (V
0
) and a peripheral sector (V
1
) [26,27]. The
V-ATPase V
1
sector contains the catalytic site which
hydrolyses ATP to translocate protons across the mem-
brane by the proton-pore forming V
0
sector. In the ER, the
assembly of the V-ATPase starts with the V
0
-sector and may
be completed in this compartment by the build-up of the V
1
onto the V
0
[28,29]. Given the pH in the ER, the V-ATPase
should be considered as being essentially inactive in this part
of the secretory pathway. An active V-ATPase is required
further downstream in the secretory pathway but it is not
known in which compartment the V-ATPase becomes
active and which mechanism is involved in the targeting of
the V-ATPase to the various secretory pathway compart-
ments. V-ATPase interacting proteins, such as the accessory
subunit Ac45, may play an important role in this targeting
process, as Ac45 has been shown to interact with the
Correspondence to G. J. M. Martens, Department of Animal Physi-
ology, Nijmegen Center for Molecular Life Sciences, University of
Nijmegen, Geert Grooteplein Zuid 28, 193RT, 6525 GA Nijmegen,
the Netherlands. Fax: + 31 24 3615317, Tel.: + 31 24 3610564,
E-mail: g.martens@ncmls.kun.nl
Abbreviations: Baf, bafilomycin A1; BFA, brefeldin A; EndoH,
endoglycosidase H; ER, endoplasmic reticulum; NDGA, nordi-
hydroguaiaretic acid; NIL, neurointermediate lobe; PC2, prohormone
convertase 2; POMC, proopiomelanocortin; TGN, trans-Golgi net-
work; V-ATPase, vacuolar H
+
-ATPase.
Note: a web page is available at
http://www.kun.nl/molanphys/Homepage/home.htm
(Received 26 October 2001, revised 6 February 2002, accepted 8
February 2002)
Eur. J. Biochem. 269, 1844–1853 (2002) ÓFEBS 2002 doi:10.1046/j.1432-1033.2002.02831.x
membrane sector of the V-ATPase in only a subset of
organelles [30]. Ac45 was initially isolated from bovine
chromaffin granules and identified as a type I transmem-
brane protein of 45 kDa [30]. However, N-terminal
sequencing of the isolated 45-kDa protein and the cloning
of full-length Ac45 cDNA revealed that the isolated protein
represents a cleaved fragment of a larger protein [30,31]. In a
differential screening strategy aimed at identifying genes
that are involved in the biosynthesis and release of peptide
hormones, we isolated a cDNA (X1311) encoding Ac45 of
the amphibian Xenopus laevis [32]. The melanotrope cells of
the Xenopus intermediate pituitary were used for this
screening approach because the activity of these neuroen-
docrine cells can be physiologically stimulated by placing
the animal on a black background. The cellular activation
results in the production and release of large amounts of the
proopiomelanocortin (POMC)-derived melanophore-stimu-
lating hormone, which causes pigment dispersion in dermal
melanophores, thereby darkening the skin [33]. Approxi-
mately 10 times more Ac45 transcripts have been found in
the melanotrope cells of animals adapted to a black
background compared to those of white-adapted animals
[32], suggesting that Ac45 has an important role in the
regulated secretory pathway of neuroendocrine cells.
Here, we examined in detail the fate of the Ac45 protein
in the melanotrope cells of Xenopus intermediate pituitary.
We found that in these cells, the folding and proteolytic
cleavage of intact Ac45 is slow, and occurs in the early
secretory pathway where activation of V-ATPases is
required.
MATERIALS AND METHODS
Animals
South-African clawed toads, Xenopus laevis,werebredand
reared in the aquarium facility of the Department of Animal
Physiology of the University of Nijmegen. Animals were
adapted to a black background by keeping them in black
buckets under constant illumination for at least three weeks
at 22 °C. All experiments were carried out under the
guidelines of the Dutch law concerning animal welfare.
Biochemicals and antibodies
Rabbit polyclonal antisera 1311C and 1311N, directed
against a synthetic peptide comprising the 12 C-terminal
amino-acid residues of Xenopus Ac45 and against a
recombinant fragment of Xenopus Ac45 (comprising ami-
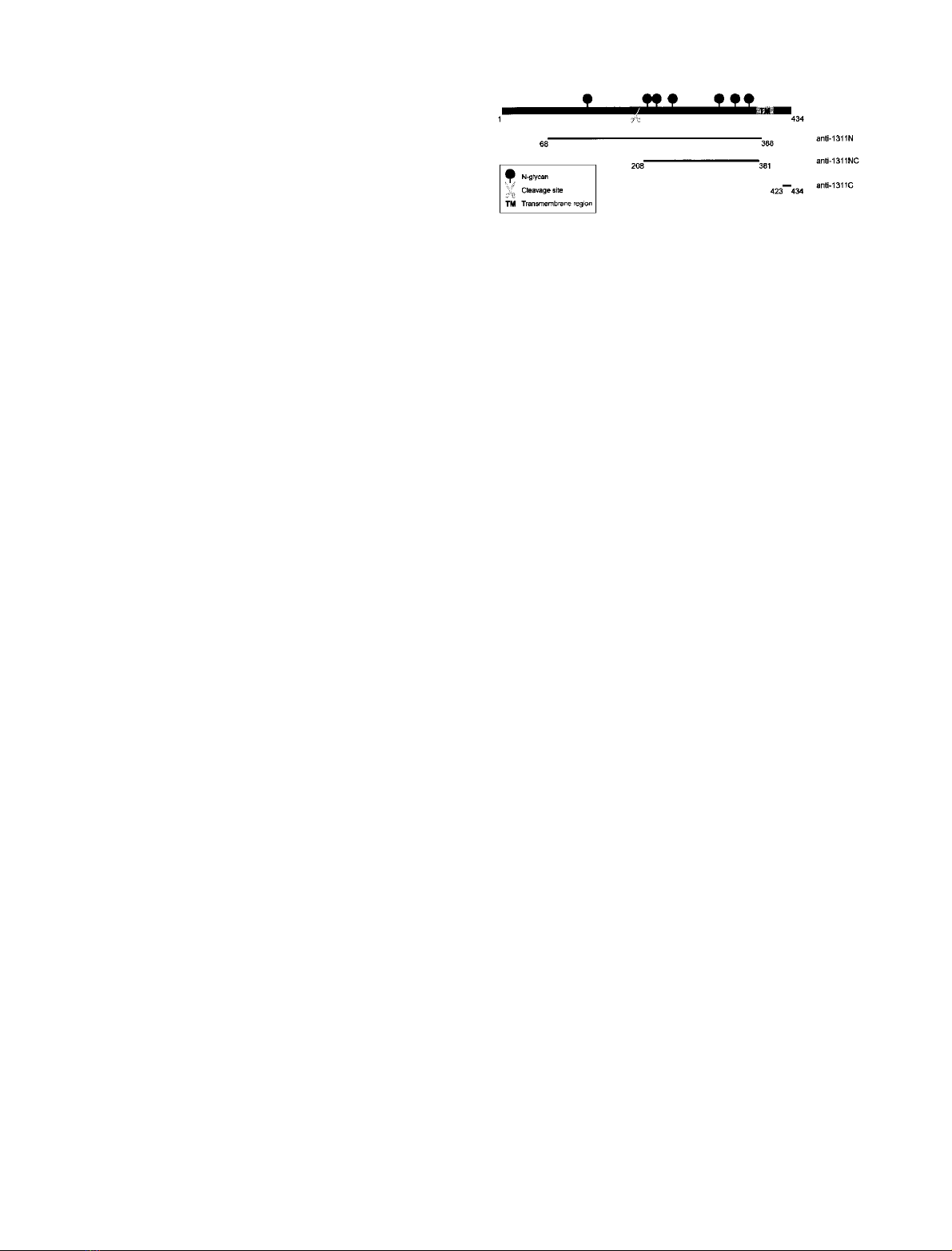
no-acid residues Gly68 to Pro388 with a hexahistidine tail at
its N-terminus; numbering according to [34]), respectively,
have been described previously [34] (Fig. 1). Rabbit poly-
clonal antiserum 1311NC was raised against a recombinant
polypeptide corresponding to amino-acid residues Pro208–
Ser381 (numbering according to [34]) of Xenopus Ac45
expressed in E. coli as a fusion protein with a hexahistidine
tag at its C-terminus (Cogon, Hilden, Germany) (Fig. 1).
Brefeldin A (BFA), monensin, nordihydroguaiaretic acid
(NDGA), chloroquine, and tunicamycin were purchased
from Sigma (St Louis, MO, USA). Leupeptin was from
Roche Diagnostics (Mannheim, Germany) and bafilomycin
A1 (Baf) from Wako Pure Chemical Industries (Osaka,
Japan).
Metabolic labeling of
Xenopus
neurointermediate lobes
and immunoprecipitation analysis
Neurointermediate lobes (NILs) from black-adapted Xen-
opus laevis were dissected and preincubated in methionine-
and cysteine-free culture medium [6.7 mL L15 medium
(Gibco-BRL, Gaithersburg, MD, USA), 3 mL milli-Q
water, 10 lgÆmL
)1
kanamycin, 1% antibiotic-antimycotic
solution (Gibco-BRL), 8 mg CaCl
2
, 3 mg bovine serum
albumin and 2 mg glucose] for 30 min at 22 °C. Pulse
labeling of newly synthesized proteins was performed by
incubating the lobes in methionine/cysteine-free culture
medium containing 5 mCiÆmL
)1
[
35
S]Met/Cys (Promix,
Amersham, Buckinghamshire, UK) for 1 h at 22 °C.
Subsequent chase incubations were in culture medium
supplemented with 5 m
ML
-methionine, 2.5 m
ML
-cysteine
and 10% fetal bovine serum. BFA (2.5 lgÆmL
)1
)was
present during the pre-, pulse and chase incubations, unless
stated otherwise. NDGA (30 l
M
) was present only during
the chase incubation. In some experiments, lobes were first
incubated overnight in the absence or presence of
10 lgÆmL
)1
tunicamycin in culture medium containing
10% fetal bovine serum (Gibco-BRL). For immunoprecipi-
tation analysis, lobes were homogenized on ice in lysis buffer
(50 m
M
Hepes pH 7.2, 140 m
M
NaCl, 10 m
M
EDTA,
1% Tween-20, 0.1% Triton X-100, 0.1% deoxycholate)
containing 1 m
M
phenylmethanesulfonyl fluoride and
0.1 mgÆmL
)1
soybean trypsin inhibitor. Homogenates were
cleared by centrifugation (10 000 g,7minat4°C), and
used for protein deglycosylation (see below), or directly
supplemented with 0.1 volume of 10% SDS and diluted
10-fold in lysis buffer before addition of anti-Ac45 antise-
rum (1 : 500 dilution). Immune complexes were precipitated
with protein-A–Sepharose (Pharmacia Biotech, Uppsala,
Sweden) and subjected to SDS/PAGE [35]. Gels were
processed for fluorography and radiolabeled proteins were
detected by autoradiography.
Immunoblotting
NILs dissected from black-adapted Xenopus laevis were
incubated overnight at 22 °C in culture medium with 10%
fetal bovine serum in the absence or presence of drugs, or
directly homogenized in lysis buffer containing 1 m
M
phenylmethanesulfonyl fluoride and 0.1 mgÆmL
)1
soybean
trypsin inhibitor. Lysates were cleared by centrifugation
(10 000 g,7 min,4 °C) and used for protein deglycosylation
(see below) or immediately denatured in sample buffer at
Fig. 1. Antigenic epitopes used to produce Ac45 region-specific antisera.
Recombinant proteins comprising residues Gly68 to Pro388 and
Pro208-Ser381, and a synthetic peptide corresponding to the 12
C-terminal amino-acid residues of Xenopus Ac45 were used to produce
rabbit polyclonal antisera 1311N, 1311C, and 1311NC, respectively.
ÓFEBS 2002 The fate of Ac45 in the secretory pathway (Eur. J. Biochem. 269) 1845
95 °C for 5 min Proteins were separated by SDS/PAGE
and electrotransferred to nitrocellulose (Schleicher &
Schuell, Dassel, Germany). Membranes were blocked and
washed with blocking buffer (100 m
M
NaCl; 100 m
M
Na
2
PO
4
; 1% Tween-20) containing 5% low-fat dry milk.
Blocking buffer with 2% low-fat dry milk was used for
further washing steps and incubations with primary and
secondary antibodies. The secondary antibody used was an
peroxidase-conjugated anti-(rabbit IgG) Ig (Sigma, St
Louis, MO, USA) at a dillution of 1 : 3000. Peroxidase
activity was detected using the Lumilight system (Roche
Diagnostics, Mannheim, Germany).
Deglycosylation of proteins
Proteins were treated with endoglycosidase H (EndoH)
(Roche Diagnostics, Mannheim, Germany) to remove high-
mannose N-glycans from glycoproteins. Lysates were boiled
for 10 min in 50 m
M
Na-citrate buffer (pH 5.5) containing
0.1% SDS, gradually cooled to RT, and incubated
overnight in the absence or presence of 40 mUÆmL
)1
EndoH at 37 °C. Proteins were deglycosylated by
N-glycosidase F (Roche Diagnostics, Mannheim, Germany)
to remove both high-mannose and complex oligosacchar-
ides. For this purpose, protein lysates were boiled for
10 min in 10 m
M
Hepes (pH 7.4) containing 0.1% SDS,
cooled down to RT, supplemented with 0.5% Nonidet P-40,
100 l
M
phenylmethanesulfonyl fluoride and 100 lgÆmL
)1
soybean trypsin inhibitor, and incubated overnight at 37 °C
with or without 5 U N-glycosidase F per mL.
RESULTS
Intact newly synthesized Ac45 is N-linked glycosylated
To study the biosynthesis of Ac45, we raised, in addition to
the previously produced antisera 1311N and 1311C (Fig. 1;
[34]), a third anti-Ac45 antiserum (1311NC; against a
recombinant protein comprising Xenopus Ac45 residues
208–388; Fig. 1). Following a 1-h pulse labeling of neuro-
intermediate lobes (NILs) from black-adapted Xenopus,the
1311N and 1311C antisera detected a newly synthesized
protein of 62 kDa and a less abundant protein of
64 kDa (Fig. 2, lanes 1 and 10). Both proteins represent
intact forms of Ac45 and vary only in the degree of N-linked
glycosylation, as deglycosylation of these radiolabeled
proteins by N-glycosidase F led to an 46-kDa protein
(Fig. 2, lanes 4 and 13). The mobility of the two intact forms
increased slightly during subsequent chase incubations of
4 h and 8 h, giving rise to products of 61 and 63 kDa
(Fig. 2, compare lane 1 with 2 and 10 with 11). This minor
shift in mobility is likely due to a change in the N-linked
sugars (possibly oligosaccharide trimming), as deglycosyla-
tion of these proteins again yielded a product of 46 kDa
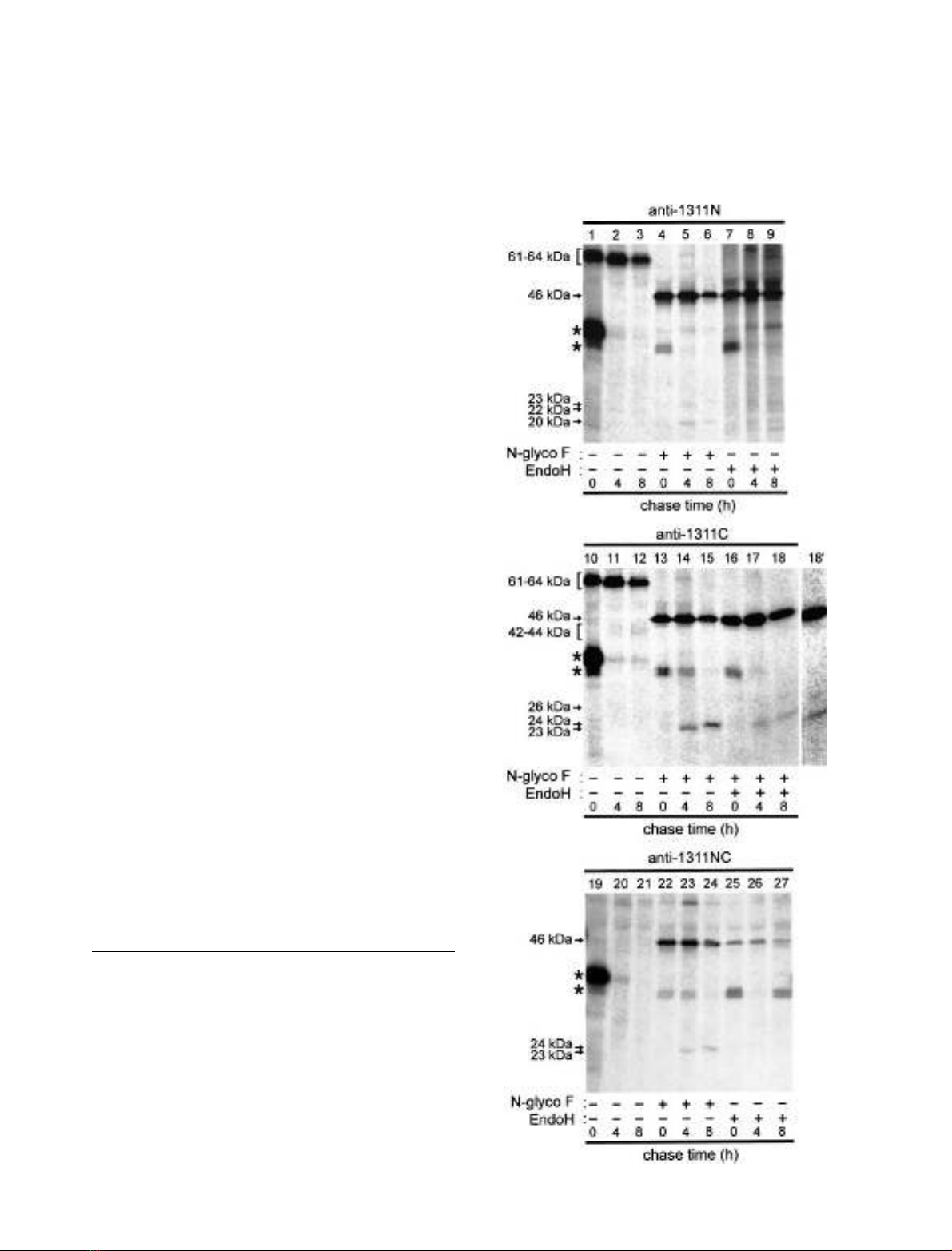
Fig. 2. Deglycosylation allows detection of the Ac45 processing products
by region-specific polyclonal antisera. NILs from black-adapted
Xenopus were pulsed for 1 h with [
35
S]Met/Cys and then chased for
the indicated time periods. Total lobe extracts were directly subjected
to immunoprecipitation with antisera 1311N, 1311C or 1311NC, or
deglycosylated by N-glycosidase F or EndoH prior to immunopre-
cipitation. Precipitated proteins were resolved by SDS/PAGE and
visualized by fluorography. Migration positions of intact and
processed forms of Ac45 are indicated. Lane 18 with increased contrast
is depicted in lane 18¢. Note that some of the immunoprecipitates
contain 37-kDa glycosylated or 35-kDa deglycosylated POMC that
bound nonspecifically (asterisk).
1846 V. Th. G. Schoonderwoert et al. (Eur. J. Biochem. 269)ÓFEBS 2002

(Fig. 2, lanes 5, 6, 14 and 15). The amount of 61- and 63-
kDa Ac45 decreased during the 8-h chase (half-life 4–6 h) as
a result of a cleavage event (see below). Both the 61- and 63-
kDa forms are not immunoprecipitated by the 1311NC
antiserum (Fig. 2, lanes 19–21). Presumably, the N-linked
glycans prevent detection of these forms as their removal by
N-glycosidase F results in the immunoprecipitation of the
deglycosylated 46-kDa intact form by this antiserum
(Fig. 2; lanes 22–24). These results show that newly
synthesized Ac45 is N-linked glycosylated to a major
product of 62 kDa and a minor product of 64 kDa
that are subsequently processed to 61- and 63-kDa
products.
Newly synthesized Ac45 is cleaved
In the melanotrope cells of the Xenopus NIL, intact
N-linked glycosylated Ac45 is intracellularly cleaved to a
C-terminal fragment of 40 kDa. Although the 40-kDa
product could be detected by Western blotting with the
C-terminally directed anti-Ac45 serum 1311C, this antise-
rum did not immunoprecipitate the newly synthesized form
of this fragment [32]. However, after optimization of the
immunoprecipitation conditions, we detected the newly
synthesized C-terminal product with antiserum 1311C as a
diffuse band of 42–44 kDa (Fig. 2, lanes 11 and 12). With
antisera 1311N and 1311NC, we could not precipitate this
product (Fig. 2, lanes 2 and 3, and 20 an 21), possibly
because of the presence of numerous N-linked glycans in
this region of the protein (Fig. 1). Indeed, after removal of
the N-linked glycans by N-glycosidase F, all three antisera
(1311N, 1311NC and 1311C) immunoprecipitated this
fragment in its deglycosylated forms, namely as proteins of
23 and 24 kDa (Fig. 2, lanes 5, 14 and 23, and 6, 15
and 24, respectively). During the chase incubation, the
mobility of the deglycosylated C-terminal fragment shifted
from 23 kDa to 24 kDa (Fig. 2, lane 14 and 15),
probably as the result of a post-translational modification.
The amount of the deglycosylated C-terminal Ac45
cleavage fragment, with a size of 23 kDa after 4 h of
chase and 24 kDa after 8 h of chase, increased during
the chase incubation (Fig. 2, lane 14 and 15), as was
expected because of the progressive cleavage of intact 61/
63-kDa Ac45. Thus, from these data, we conclude that
newly synthesized 61/63-kDaAc45iscleavedto
C-terminal products of 42–44 kDa (with deglycosylated
forms of 23 and 24 kDa).
Identification of the N-terminal Ac45 cleavage product
In contrast to what holds for the C-terminal cleavage
fragment of Ac45 [30,34], the N-terminal cleavage fragment
has not been identified yet. However, after optimization of
the immunoprecipitation conditions, from newly synthe-
sized Xenopus NIL proteins we precipitated with antiserum
1311N a low-abundant product of 22 kDa (Fig. 2, lanes
1–3). Because of the following we conclude that this 22-
kDa product is the glycosylated form of the N-terminal
Ac45 cleavage fragment. First, the size of this product is in
line with the predicted size of the N-terminal fragment that
remains following cleavage of intact 61/63-kDa Ac45 to the
42–44-kDa C-terminal product. Furthermore, both the
22-kDa fragment and its deglycosylated 20-kDa form
(see below) are not immunoprecipitated with the two
antisera raised against more C-terminally located regions of
Ac45 (1311C and 1311NC, Figs 1 and 2, lanes 10–15 and
19–24). Finally, the N-terminal Ac45 fragment contains one
potential N-linked glycosylation site (Asn128; numbering
according to [34]), and this site appears to be used, as
N-glycosidase F treatment of the NIL lysate prior to immu-
noprecipitation causes a shift in the mobility of the 22-kDa
product to 20 kDa (Fig. 2, lanes 4–6). The amount of the
N-terminal fragment would be expected to increase during
the chase period because of the progressive cleavage of
intact Ac45. However, during the chase incubation a
decrease in the level of the N-terminal fragment was found,
suggesting that this cleavage product may be subjected to an
intracellular degradation process. This circumstance may
also explain why the N-terminal Ac45 fragment has not
been detectable by Western blotting [34].
Transport of newly synthesized Ac45 to the Golgi
In the medial Golgi, N-linked oligosaccharides can be
modified to two broad classes, namely complex oligosac-
charides and high-mannose oligosaccharides. Both types
of oligosaccharides can be removed completely from
proteins by treating them with N-glycosidase F. In contrast,
endoglycosidase H (EndoH) removes only high-mannose
oligosaccharides. The acquisition of resistance of an
N-glycosylated protein to EndoH, which requires the action
of glycosylation enzymes localized in the medial Golgi, can
thus be used to determine whether a glycosylated protein
has entered the medial compartment [36]. We determined
whether the intact or the cleavage products of Ac45 acquire
resistance to digestion with EndoH. Extracts of pulse-
chased NILs were subjected to EndoH before immunopre-
cipitation with antisera 1311N, 1311C or 1311NC. All
three anti-Ac45 antisera immunoprecipitated from the
EndoH-treated NIL lysate a newly synthesized product of
46 kDa. This product corresponds with the intact newly
synthesized deglycosylated Ac45 protein that was immuno-
precipitated from NIL lysates that were treated with
N-glycosidase F, indicating that intact Ac45 is sensitive to
EndoH. This finding implies that intact Ac45 is cleaved in a
compartment before the medial Golgi. Antisera 1311N and
1311C immunoprecipitated from the EndoH-treated and
the N-glycosidase F-treated lysates similar amounts of the
46-kDa product (Fig. 2, lanes 7–9 and 16–18). In
contrast, the 1311NC antiserum precipitated a considerably
lower amount of this product from the EndoH-treated than
from the N-glycosidase F-treated lysates (Fig. 2, compare
lanes 22–24 with 25–27). Probably, the presence of the
N-acetylglucosamine residues remaining after EndoH
digestion [37], but removed by N-glycosidase F [38], lowers
the affinity of the Ac45 product for the 1311NC antiserum.
This possibility may also explain why this antiserum was
not able to detect significant amounts of the 23-kDa
C-terminal cleavage product in EndoH-treated lysates
(Fig. 2, lanes 17 and 18).
In addition to the 23-kDa product, the 1311C anti-
serum detected also a low-abundant product of 26-kDa in
the EndoH-treated lysate (Fig. 2, lanes 17 and 18/18¢). This
product was not detected in the N-glycosidase F-treated
lysate (Fig. 2, lane 15), indicating that it represents a
C-terminal Ac45 cleavage form of which most, but not all,
ÓFEBS 2002 The fate of Ac45 in the secretory pathway (Eur. J. Biochem. 269) 1847
N-glycans are sensitive to EndoH. The amount of the
23-kDa product in the EndoH-treated lysates remained
constant during the chase, whereas the analysis of the
N-glycosidase F-treated samples clearly indicated an
increase in the total amount of this fragment (Fig. 2,
compare lanes 14 and 15 with 17 and 18). These findings
suggest that at first, all the N-linked sugars on the
C-terminal cleavage product are sensitive to EndoH
(EndoH treatment gives an 23-kDa product), and that
during the chase some of the N-glycans on the C-terminal
cleavage product become resistant to EndoH (resulting in
an 26-kDa product). The N-linked sugar on the
N-terminal cleavage product also acquired resistance to
EndoH, as we found a faint band of 22 kDa in the
EndoH-treated extracts that is absent in the total lysates
of these samples (Fig. 2, lanes 8 and 9, and data not
shown).
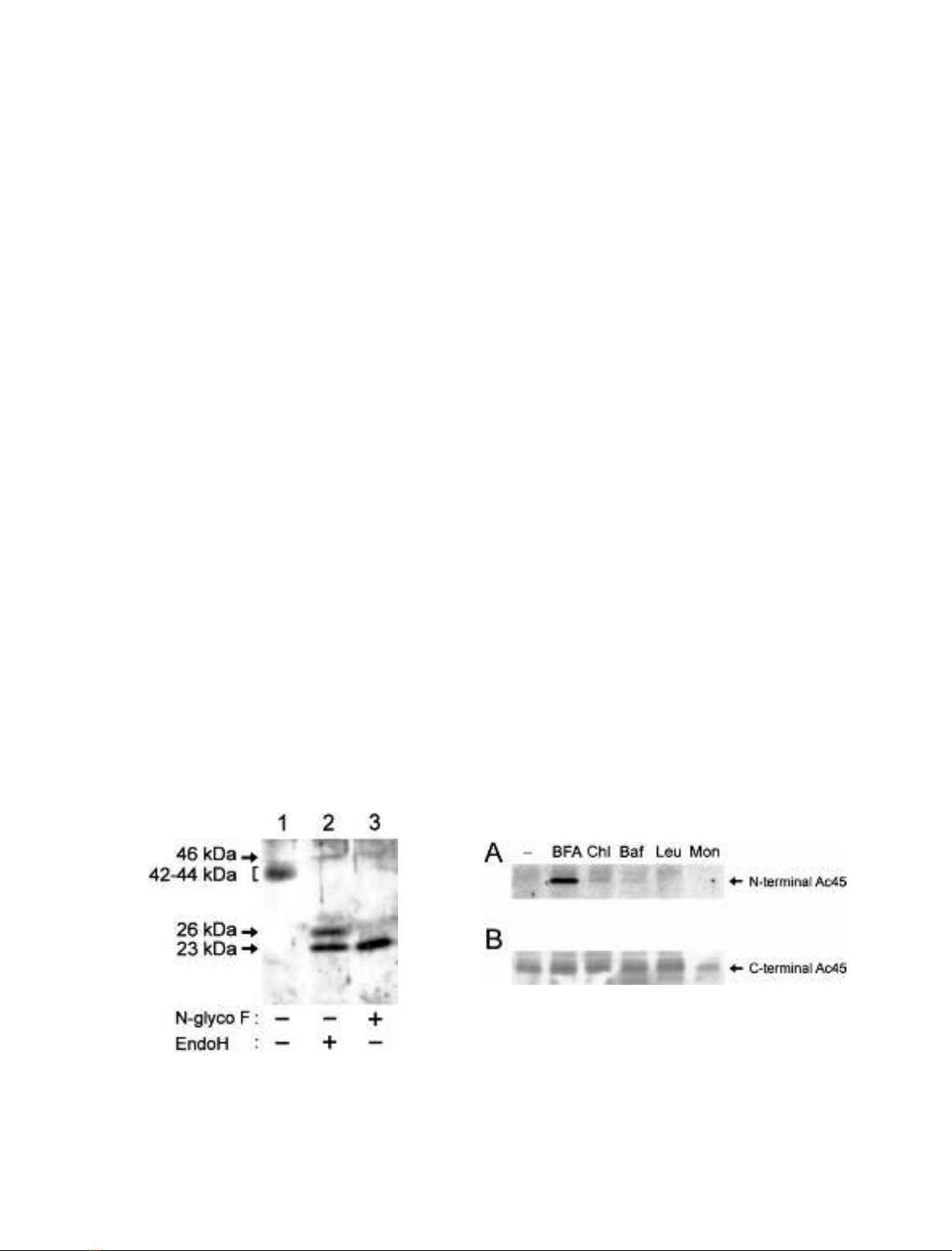
Western blot analysis was employed to study the steady
state levels of EndoH-sensitive and EndoH-resistant forms
of Ac45. In line with the results of biosynthetic studies,
EndoH treatment of the NIL lysate prior to Western blot
analysis with the 1311C antibody again resulted in the
detection of an 23-kDa and an 26-kDa product
(Fig.3,lane2).Theintensityofthe23-kDa band is
higher than that of the 26-kDa band, indicating that in
the steady state situation the 23-kDa product is the
major form in the EndoH-treated lysate. As expected,
deglycosylation by N-glycosidase F resulted in the detec-
tion of the 23-kDa C-terminal cleavage product (Fig. 3,
lane 3). As this product is more abundant in the EndoH-
treated NIL lysate than the 26-kDa product, we
conclude that at steady state, most of the glycosylated
42–44-kDa C-terminal cleavage products contain N-linked
glycans that are sensitive to EndoH.
Together, these results demonstrate that the cleavage of
intact 61/63-kDa Ac45 occurs before the medial Golgi,
and that in this compartment the N-glycan on the
N-terminal and some of the N-glycans on the C-terminal
cleavage product are converted to complex oligosaccha-
rides.
BFA inhibits the degradation of the N-terminal Ac45
fragment
As the N-terminal fragment was not detected by immu-
noblotting, we hypothesized that it may be degraded
intracellularly. We examined this possibility by affecting
Ac45 transport through the secretory pathway via drugs
that interfere with intracellular protein transport, namely
the fungal metabolite brefeldin A (BFA) and the sodium
ionophore monensin. BFA causes fusion of Golgi mem-
branes with the ER and the retention of newly synthesized
proteins in a lumenal milieu characteristic of the early
compartments of the secretory pathway [39]. In addition,
BFA blocks the exit of proteins from the TGN [40] as we
have recently shown for several regulated secretory proteins
in Xenopus melanotrope cells [41]. Monensin interferes with
protein transport between Golgi compartments [42]. Fur-
thermore, to examine if the N-terminal cleavage product is
degraded in lysosomes, a number of compounds that
interfere with lysosomal function were used. Leupeptin is a
thiol protease inhibitor that inhibits degradation of proteins
in lysosomes [43]. The weak base chloroquine and the
V-ATPase-specific inhibitor bafilomycin A1 (Baf) are
known to inhibit lysosomal and endosomal enzymes by
disturbing the intralumenal pH [1]. Baf may also affect the
transport of intact Ac45 or its cleavage products in a post-
TGN compartment, e.g. in Xenopus melanotrope cells [41].
To examine the effects of the above-mentioned drugs,
Xenopus NILs were incubated overnight in the absence or
presence of a drug, and the lobes were lysed and subjected to
Western blotting with the 1311N or 1311C antiserum. The
N-terminal cleavage fragment of Ac45 did not accumulate
when NILs were incubated in the presence of monensin or
the lysosomal inhibitors leupeptin, chloroquine and Baf.
However, in NILs incubated in the presence of BFA, an
22-kDa product had clearly accumulated (Fig. 4A). This
product could be deglycosylated with N-glycosidase F to
20 kDa and was not detected with the 1311C antibody
(data not shown), indicating that this product represents
the N-terminal fragment of Ac45. The drugs used did
not significantly change the amount of the 42–44-kDa
Fig. 4. Effect of inhibitors of intracellular transport and lysosomal
function on the degradation of the N-terminal 22-kDa Ac45 cleavage
fragment. NILs dissected from black-adapted Xenopus were incubated
overnight in medium with no drugs, brefeldin A (BFA, 2.5 lgÆmL
)1
),
chloroquine (Chl, 100 l
M
), bafilomycin A1 (Baf, 1 l
M
), leupeptin
(Leu, 100 lgÆmL
)1
), or monensin (Mon, 100 n
M
). Proteins were
extracted from these NILs, separated by SDS/PAGE, transferred to
nitrocellulose and probed with the anti-Ac45 serum 1311N to detect
the 22-kDa N-terminal fragment (A) or 1311C to detect the 42 to
44-kDa C-terminal fragment (B).
Fig. 3. Steady-state levels of EndoH-sensitive and -resistant forms of the
C-terminal Ac45 cleavage fragment. Total NIL extracts from black-
adapted Xenopus were incubated overnight with no enzyme (lane 1),
EndoH (lane 2), or N-glycosidase F (lane 3). Reactions were stopped
by adding SDS sample buffer, and the samples were subjected to SDS/
PAGE and immunoblotting, using 1311C.
1848 V. Th. G. Schoonderwoert et al. (Eur. J. Biochem. 269)ÓFEBS 2002


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)