
Intrinsic disorder and coiled-coil formation in prostate
apoptosis response factor 4
David S. Libich
1,
*, Martin Schwalbe
1,
*, Sachin Kate
1
, Hariprasad Venugopal
1
, Jolyon K. Claridge
1
,
Patrick J. B. Edwards
1
, Kaushik Dutta
2
and Steven M. Pascal
1
1 Centre for Structural Biology, Institute of Fundamental Sciences and Department of Physics, Massey University, Palmerston North,
New Zealand
2 New York Structural Biology Centre, NY, USA
Introduction
Prostate apoptosis response factor-4 (Par-4) is an ubi-
quitously expressed and evolutionary conserved protein
that was initially identified as a pro-apoptotic factor in
rat AT-3 androgen-independent prostate cancer cells
exposed to ionomycin [1,2]. The identified pro-apopto-
tic and tumour-suppressive roles of Par-4 are consid-
ered to be its most important cellular functions and,
accordingly, Par-4 is downregulated in various cancers
[3]. The anti-cancer strategy employed by Par-4 is
achieved by direct activation of the cell-death machinery
(e.g. Fas ⁄FasL) [4] and inhibition of pro-survival fac-
tors (e.g. nuclear factor-kappa B) [5]. Furthermore,
ectopic over-expression of Par-4 can either directly
induce apoptosis or sensitize cancer cells to apoptotic
stimuli, dependent on cell type [6]. Primarily a cyto-
plasmic protein, translocation of Par-4 to the nucleus
is linked with the direct induction of apoptosis in
cancer cells [3,7,8]. Initially characterized in prostate
cancer, Par-4 has also been demonstrated to function
in renal cell carcinomas [9], leukaemia [10] and
Keywords
circular dichroism; coiled-coil; intrinsically
disordered protein; prostate apoptosis
response factor 4; solution NMR
spectroscopy
Correspondence
D. S. Libich or S. M. Pascal, Institute of
Fundamental Sciences, Massey University,
Turitea Site, Private Bag 11222, Palmerston
North 4442, New Zealand
Fax: +64 6 350 5682
Tel: +64 6 356 9099
E-mails: d.s.libich@massey.ac.nz;
s.pascal@massey.ac.nz
*These authors contributed equally to this
work
(Received 24 March 2009, accepted 6 May
2009)
doi:10.1111/j.1742-4658.2009.07087.x
Prostate apoptosis response factor-4 (Par-4) is an ubiquitously expressed
pro-apoptotic and tumour suppressive protein that can both activate cell-
death mechanisms and inhibit pro-survival factors. Par-4 contains a highly
conserved coiled-coil region that serves as the primary recognition domain
for a large number of binding partners. Par-4 is also tightly regulated by
the aforementioned binding partners and by post-translational modifica-
tions. Biophysical data obtained in the present study indicate that Par-4
primarily comprises an intrinsically disordered protein. Bioinformatic
analysis of the highly conserved Par-4 reveals low sequence complexity and
enrichment in polar and charged amino acids. The high proteolytic suscep-
tibility and an increased hydrodynamic radius are consistent with a largely
extended structure in solution. Spectroscopic measurements using CD and
NMR also reveal characteristic features of intrinsic disorder. Under physio-
logical conditions, the data obtained show that Par-4 self-associates via the
C-terminal domain, forming a coiled-coil. Interruption of self-association
by urea also resulted in loss of secondary structure. These results are
consistent with the stabilization of the coiled-coil motif through an intra-
molecular association.
Abbreviations
CREB, cAMP-responsive element-binding protein; DLS, dynamic light scattering; GST, glutathione S-transferase; HSQC, heteronuclear single
quantum coherence; IDP, intrinsically disordered protein; IPTG, isopropyl thio-b-D-galactoside; LZ, leucine zipper; NLS, nuclear localization
sequence; Par-4, prostate apoptosis response factor 4; PK, protein kinase; SAC, selective apoptosis of cancer cells.
3710 FEBS Journal 276 (2009) 3710–3728 ª2009 The Authors Journal compilation ª2009 FEBS

neuroblastomas [11], as well as endometrial [12],
pancreatic [13] and gastric [8] cancers.
In addition to its role in cancer, Par-4 is thought to
assist in normal neuronal development by preventing
the hyper-proliferation of nerve tissues, in turn con-
trolling the number of neurones and glial cells in both
the peripheral and central nervous systems [14,15].
Par-4 is upregulated in several neurodegenerative dis-
eases, such as Alzheimer’s disease [16,17], Parkinson’s
disease [18], Huntington’s disease [19] and amyotrophic
lateral sclerosis [20]. Par-4 is also reportedly involved
in immune response modulation [21], synaptic function
modulation [22] and apoptosis of neurones that have
received a traumatic insult [23].
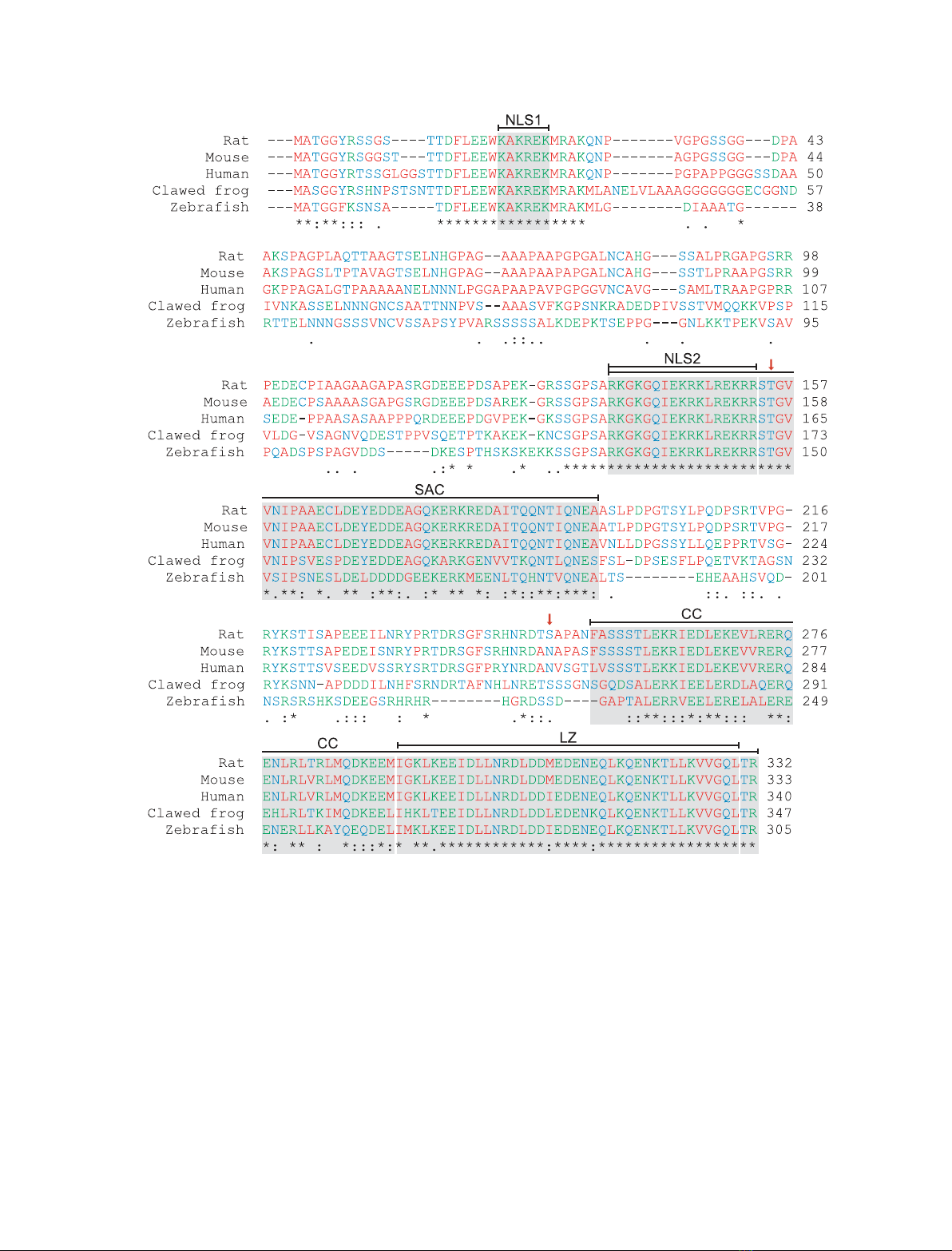
The C-terminal quarter of Par-4 (Fig. 1) is highly
conserved and shares some homology with the death
domains of other apoptotic proteins, such as Fas,
receptor-interacting protein, Fas-associated death
domain protein and tumour necrosis factor receptor-
associated death domain protein [24,25]. This region
functions as the primary recognition and binding site
for various partners of Par-4, including Wilms’ tumour
1 [7], Akt1 ⁄protein kinase (PK) B [26], atypical PKCs
(PKCs fand k⁄i) [24], p62 [27], death-associated pro-
tein-like ⁄zipper interacting kinase [28], THAP [29],
Amida [30], E2F1 [31], D
2
dopamine receptor [32],
b-site amyloid precursor protein cleaving enzyme 1
[17], apoptosis-antagonizing transcription factor [33]
and topoisomerase 1 [34]. In addition, several binding
partners have been shown to interact at various sites
N-terminal to the aforementioned C-terminal segment,
including the androgen receptor [35], F-actin [36],
14-3-3 [26] and the SPRY domain-containing suppressor
of cytokine signalling box proteins 1, 2 and 4 [37].
Par-4 contains several conserved phosphorylation
sites that are modified by kinases, such as PKA, PKC,
casein kinase II and Akt1, adding a further level of
regulation of the function of Par-4 [38]. Phosphoryla-
tion of an absolutely conserved threonine (rat T155,
human T163 or mouse T156; Fig. 1) by PKA is
required for nuclear translocation [8]. Phosphorylation
of a C-terminal serine residue (rat S249, human or
mouse S231; Fig. 1) by Akt1 effectively inactivates
Par-4 by allowing the chaperone protein 14-3-3 to bind
and sequester it in the cytoplasm, even if it is
phosphorylated on T155 [26].
These multiple interactions coupled with a high
degree of sequence conservation and post-translational
modification suggest that the in vivo role(s) of Par-4
are highly temporally and spatially regulated. Simi-
larly, the ubiquitous expression, post-translational
modifications and a plethora of binding partners are
characteristics common to many intrinsically disor-
dered proteins (IDPs) [39]. In the present study, we
demonstrate that residual structure exists in Par-4
because the measured hydrodynamic radius increased
under denaturing conditions, suggesting that the
ensemble becomes less compact. CD and NMR indi-
cate that Par-4 is primarily intrinsically disordered
under physiological conditions and exists as an ensem-
ble of fast-averaging (on the NMR time-scale) struc-
tures. Furthermore, Par-4 forms a stable coiled-coil
through a self-association event mediated by the C-ter-
minus. The coiled-coil was probed using increasing
concentrations of chaotropic agents and was found to
be very stable. Using NMR, the segment of Par-4 not
involved in the coiled-coil was shown to have spectral
features that were similar to those of a C-terminal
deletion mutant. This is important because it suggests
that Par-4 is able to bind more than one partner at a
time and thus could function as a hub linking the
functions of several proteins. The coiled-coil region of
Par-4 represents an important functional domain that
is an example of a gain of structure upon binding tran-
sition, which is another important feature of IDPs [40].
Results
All sequence numbering is made with reference to rat
Par-4, to reflect the recombinant rat (rrPar-4) constructs
used in these studies. Three constructs were created;
rrPar-4FL (Par-4 full-length, residues 1–332), rrPar-
4DLZ (deleted leucine zipper, residues 1–290) and rrPar-
4SAC [selective apoptosis of cancer cells (SAC) domain
construct, residues 137–195] (Fig. 2A). The sequence
identity expressed relative to rat Par-4 of mouse and
human is 92% and 76%, respectively, whereas African
clawed frog and zebra fish share 52% and 47%
sequence identity with rat, respectively (Fig. 1).
The nuclear localization sequences (NLS) 1 (residues
20–25) and 2 (residues 137–153) are strictly conserved
in all known Par-4 sequences (Fig. 1). The SAC
domain, which includes NLS2, is the minimum frag-
ment of Par-4 that is absolutely required for apoptosis
[6] and is completely conserved amongst mammals
(Fig. 1). Furthermore, there is a high degree of
sequence conservation in the C-terminal quarter of Par-
4, which contains primarily a coiled-coil-like sequence
(residues 254–332; Figs 1 and 2A). In particular, a leu-
cine zipper (residues 292–330), which is a subset of the
coiled-coil domain, is almost conserved in all known
Par-4 sequences, suggesting a common functionality
(Figs 1 and 2A). Relatively few Par-4 genes have been
sequenced. It has been suggested that the general pat-
tern of sequence conservation shown in Fig. 1 is likely
to be conserved across other mammalian sequences [1].
D. S. Libich et al. Intrinsic disorder in Par-4
FEBS Journal 276 (2009) 3710–3728 ª2009 The Authors Journal compilation ª2009 FEBS 3711
Based on disembl analysis [41], the majority (> 70%)
of Par-4 is predicted to be disordered. The putative
regions of order in Par-4, as indicated by grey bars in a
disembl plot (Fig. 2B), align with or occur within func-
tionally important regions of Par-4 (Fig. 2A), namely
NLS1, NLS2, SAC and the coiled-coil. Secondary
Fig. 1. Sequence alignment of the prostate apoptosis response factor 4 (Par-4). A BLASTP ⁄CLUSTALW [102,103] alignment of sequences of
Par-4 from various species: rat (Rattus norvegicus), mouse (Mus musculus), human (Homo sapiens), African clawed frog (Xenopus laevis)
and zebra fish (Danio rero). The amino acids are coloured: red (nonpolar side chains: G, A, V, L, I, M, P, F and W), blue (polar side chains: S,
T, N, Q, Y and C) and green (polar, charged side chains: K, R, H, D and E). Symbols: residues in that column are identical in all sequences
(*); substitutions are conservative (:); and substitutions are semi-conservative (.). The high degree of sequence conservation of Par-4
suggests functional significance and thus resistance to evolutionary pressure. With reference to the numbering of rat Par-4, several seg-
ments are of notable interest: two nuclear localization sequences [NLS1 (20–25) and 2 (137–153)], which are completely conserved among
all known Par-4s, and the SAC domain (137–195), which is defined by being the absolute minimum fragment required for apoptosis and
includes NLS2 [6]. The C-terminal domain (254–332) is a coiled-coil (CC) motif that encompasses a LZ (292–330) as a subset. Two important
phosphorylation sites, T155 and S249, are denoted by red arrows.
Intrinsic disorder in Par-4 D. S. Libich et al.
3712 FEBS Journal 276 (2009) 3710–3728 ª2009 The Authors Journal compilation ª2009 FEBS

structure prediction using gor4 [42] shows that the
regions with the highest helical propensity also occur in
the aforementioned regions and align with the disembl
predicted ordered regions (Fig. 2C). The hydrophobic
cluster analysis [43] of Fig. 2D indicates that the most
hydrophobic regions align with the putative ordered and
predicted helical regions.
A plot of mean net charge against mean hydropho-
bicity determined from a protein’s primary structure
may be used to classify it as folded or intrinsically dis-
ordered. Plot space is divided by an empirically deter-
mined line (
R¼2:785
H1:151) based on an analysis
by Uversky et al. [44]. The three constructs used in this
study are plotted in Fig. 3A along with several ‘classi-
cally folded’ proteins. Here, rrPar-4FL, rrPar-4DLZ
and rrPar-4SAC clearly fall into disordered space gen-
erally characterized by low mean hydrophobicity and
high net charge. The construct representing the SAC
domain (rrPar-4SAC), with 14 positively charged and
13 negatively charged residues but few hydrophobic
residues, lies further in the disordered region.
Figure 3B describes the sequence complexity of
rrPar-4FL by comparison with the percent difference
between the amino acid usage of a set of known IDPs
Fig. 2. (A) A block diagram of the three constructs of rrPar-4 used in the present study. Marked on each construct are the primary regions
of functional importance, including the nuclear localization sequences [NLS1 (20–25) and 2 (137–153), coloured green], the region necessary
for SAC (137–195), the coiled-coil C-terminal domain (CC, 254-332, coloured red) and the LZ (292–330, shown with hatching). The rrPar-
4DLZ construct lacks residues 291–332, which is approximately one-half of the coiled-coil and the entire leucine zipper. The rrPar-4SAC con-
struct represents residues 137–195 of Par-4, including NLS2. All three constructs used in the present study have an N-terminal GGS tag, a
remnant from the cleavage of the purification tag, which is omitted here for simplicity. (B) DISEMBL predicts regions of order ⁄disorder in pro-
teins using neural networks trained on multiple definitions of disorder [41]. The dashed line in (B) represents a threshold value separating
order and disorder. (C) Secondary structure (a-helix only shown) prediction using GOR4 [42] and (D) hydrophobic cluster analysis (HCA) [43], a
visually enhanced representation of the primary sequence that highlights clustering of hydrophobic residues using symbols ( ,T; ,S;¤,G;
w, P) and colours (red: P and acidic residues D, E, N, Q; blue: basic residues, H, K, R; green: hydrophobic residues, V, L, I, F, W, M, Y;
black: all other residues, G, S, T, C, A). The grey bars indicate the predicted regions of order in (B) and, for comparison, are extended over
(C) and (D).
D. S. Libich et al. Intrinsic disorder in Par-4
FEBS Journal 276 (2009) 3710–3728 ª2009 The Authors Journal compilation ª2009 FEBS 3713

versus a set of folded proteins (black bars). Positive
values indicate a depletion, whereas negative bars indi-
cate an enrichment relative to folded proteins. The pat-
tern of amino acid usage for rrPar-4FL (grey bars) is
in accordance with that generally observed for IDPs
[45,46], namely a depletion of order-promoting amino
acids (L, N, F, Y, I, W, C) and enrichment of dis-
order-promoting residues (S, Q, K, P, E). The amino
acid usage for rrPar-4DLZ and rrPar-4SAC follows a
similar pattern (not shown).
As calculated (i.e. from sequence) and experimen-
tally determined [i.e. from MS, Tricine-PAGE and
dynamic light scattering (DLS)], the molecular weights
for rrPar-4FL, rrPar-4DLZ and rrPar-4SAC are given
in Table 1. Because DLS measures the Stokes radius
(R
S
) of a particle, the equation log(R
S
) = 0.357 ·
log(MW) )0.204 was used to convert R
S
to MW for
comparative purposes [47,48]. Although this approxi-
mate calculation does not take into account the shape
of the particle (i.e. it assumes a sphere), the result is
useful for illustrating the degree of extended structure
in the protein.
The primary structure predicts MWs of 36.1, 31.1 and
7.0 kDa for rrPar-4FL, rrPar-4DLZ and rrPar-4SAC,
respectively. MALDI-TOF mass spectroscopy was used
to assess the purity and determine the sizes of the con-
structs produced. The sizes determined for rrPar-4DLZ
(44.5 Da difference between expected and observed) and
rrPar-4SAC (6.6 Da difference between expected and
observed after accounting for
15
N labelling of the sample
used for MS analysis) agree within error (approximately
0.1%) with the sizes predicted from sequence analysis
(Table 1). MS revealed that the rrPar-4FL construct is
approximately 0.2 kDa larger than expected.
Relative mobility analysis of the electrophoretic pro-
files of rrPar-4FL, rrPar-4DLZ and rrPar-4SAC using
a denaturing Tricine-PAGE system (see Experimental
procedures) determined apparent molecular weights of
49.1, 41.5 and 12.4 kDa, respectively. These sizes are
significantly larger (36%, 33% and 77% larger for
rrPar-4FL, rrPar-4DLZ and rrPar-4SAC, respectively)
than the expected MWs determined from the primary
structure or MS (Table 1).
The results of DLS experiments are shown in Table 2
and summarized in Table 1. The measured R
S
for
rrPar-4FL was 189 A
˚, which is much larger than
expected for a monomeric random coil, suggesting a
polymeric state for rrPar-4FL under these conditions.
Fig. 3. (A) Charge ⁄hydrophobicity plot of rrPar-4FL (335 residues),
rrPar-4DLZ (293 residues), and rrPar-4SAC (61 residues). The divid-
ing line
R¼2:785
H1:151 represents an empirically determined
divisor between intrinsically disordered (high charge, low hydropho-
bicity) and structured (low charge, high hydrophobicity) space.
Proteins such as aprotinin [104], actin [105], ubiquitin [106] and 3C
protease [107] are plotted as examples of classically folded
proteins. (B) Sequence complexity of rrPar-4FL (grey bars) com-
pared with the average amino acid distribution of IDPs (black bars)
relative to the average amino acid distribution of globular proteins.
The relative distributions were sampled from proteins (both IDPs
and folded) deposited in the Protein Data Bank. Positive and nega-
tive values indicate an enrichment or depletion, respectively, of a
particular residue relative to globular proteins. Residues marked
with an asterisk occur two-fold more or less frequently, on average,
in IDPs than in globular proteins [46].
Table 1. Hydrodynamic properties of rrPar-4 constructs using vari-
ous biophysical techniques. MW (kDa) and hydrodynamic radius (A
˚)
are shown in the format MW (R
S
) for three constructs using four
techniques. R
S
and MW were calculated from the primary structure
in reference to a folded conformation using log(R
S
) = 0.357 ·
log(MW) )0.204.
Construct Sequence
Method of analysis
MS PAGE DLS
rrPar-4FL 36.1 (26.5) 36.2 (26.5) 49.5 (29.6) 8899 (189)
rrPar-4-DLZ 31.1 (25.1) 31.2 (25.1) 41.5 (27.8) 64.1 (32.5)
rrPar-4 SAC 7.0 (14.8) 7.1 (14.8) 12.5 (18.1) 18.7 (20.9)
Intrinsic disorder in Par-4 D. S. Libich et al.
3714 FEBS Journal 276 (2009) 3710–3728 ª2009 The Authors Journal compilation ª2009 FEBS







![Vaccine và ứng dụng: Bài tiểu luận [chuẩn SEO]](https://cdn.tailieu.vn/images/document/thumbnail/2016/20160519/3008140018/135x160/652005293.jpg)

















