
MINIREVIEW
Barrier passage and protein dynamics in enzymatically catalyzed
reactions
Dimitri Antoniou
1
, Stavros Caratzoulas
1,
*, C. Kalyanaraman
1
, Joshua S. Mincer
1
and Steven D. Schwartz
1,2
1
Department of Biophysics, Albert Einstein College of Medicine, Bronx, NY, USA;
2
Department of Biochemistry, Albert Einstein
College of Medicine, Bronx, NY, USA
This review describes studies of particular enzymatically
catalyzed reactions to investigate the possibility that catalysis
is mediated by protein dynamics. That is, evolution has
crafted the protein backbone of the enzyme to direct vibra-
tions in such a fashion to speed reaction. The review presents
the theoretical approach we have used to investigate this
problem, but it is designed for the nonspecialist. The results
show that in alcohol dehydrogenase, dynamic protein
motion is in fact strongly coupled to chemical reaction in
such a way as to promote catalysis. This result is in concert
with both experimental data and interpretations for this and
other enzyme systems studied in the laboratories of the two
other investigators who have published reviews in this issue.
Keywords: protein dynamics; enzyme catalysis; tunneling;
promoting vibration; promoting mode.
INTRODUCTION
The transmission of an atom or group of atoms from the
reactant region of a reaction to the product region under the
control of an enzyme is central to biochemistry. The manner
in which the enzyme speeds this transfer is in some cases still
not clear. What is known is the end effect; enzymatic
reactions occur at rates many orders of magnitude more
rapid than the corresponding solution phase reactions. This
review will describe work recently completed in our group
that has focused on examining the possibility that protein
dynamics may in some enzymes play a central role in helping
to produce the catalytic effect. These types of motions,
which we have termed rate promoting vibrations,are
motions of the protein matrix that change the geometry of
the chemical barrier to reaction. By this we mean that both
the height and width of the barrier are changed. This unique
role for the protein matrix has significant implications for
the dynamics of the chemical reaction; in particular, causing
a barrier to narrow can significantly enhance a light
particle’s ability to tunnel, while masking the normal kinetic
indicators of such a phenomenon. It is this feature that we
have proposed as a unifying principle for some experimental
data relating to tunneling in enzymatic reactions.
This paper will describe our studies of rate promoting
vibrations in enzymatic reactions with particular attention
to the physical origins of the phenomenon. The structure of
this paper will be as follows: in the next section, we will
briefly review a number of different potential mechanisms
for enzyme catalytic action along with promoting vibra-
tions. Following this, we will describe the mathematical
foundation for our theories in some detail. This section will
be written for nonexperts, but will contain the necessary
formulae for the specialist as well. It will include the
relationship between the current theories and a well-known
approach to charged particle transfer in biological reactions,
namely the Marcus theory. In this section we will also
describe a simple nonbiological chemical system in which
the physical features of promoting vibrations may be easily
understood – proton transfer in organic acid crystals. We
will then describe how we have used these concepts to fit
seemingly anomalous kinetic data for enzymatic reactions.
In the next section, we explore how one might rigorously
identify the presence of such a promoting vibration in any
enzymatic reaction, and illustrate the concepts with appli-
cations to specific enzyme systems. The paper then con-
cludes with discussions of future directions for this research.
POTENTIAL MODES OF ENZYMATIC
ACTION
The exact physical mechanisms by which enzymes cause
catalysis is still a topic for vigorous dialogue [1–3]. The
research described in this paper will argue for a strong
contribution from a nontraditional source, i.e. directed
protein motions. In order to place this concept into a
context, we will briefly review other potential mechanisms
for enzymes to cause catalysis. We emphasize that none of
these mechanisms are mutually exclusive, and are probably
all involved in catalysis to a greater or lesser extent in each
enzyme system.
One of the earliest and still widely accepted ideas used to
explain this catalytic efficiency is the transition state-binding
concept of Pauling [4]. In this picture, as a chemical
substance is being transformed from reactants to products,
the species that binds most strongly to the enzyme is at some
Correspondence to S. D. Schwartz, 1300 Morris Park Ave.,
Bronx, NY 10461, USA. Tel.: + 1 718 430 2139,
E-mail: sschwartz@aecom.yu.edu
Abbreviations: NAC, near attack conformations; HLADH, horse liver
alcohol dehydrogenase; YADH, yeast alcohol dehydrogenase.
Note: a website is available at http://www.aecom.yu.edu/home/sggd/
faculty/schwartz.htm
*Present address: Department of Chemical Engineering, Princeton
University, NJ, USA.
(Received 8 March 2002, revised 31 May 2002, accepted 6 June 2002)
Eur. J. Biochem. 269, 3103–3112 (2002) FEBS 2002 doi:10.1046/j.1432-1033.2002.03021.x

intermediate point thought to be at or near the top of the
solution phase (i.e. uncatalyzed) barrier to reaction. This
preferential binding releases energy that stabilizes the
transition state and thus lowers the barrier to reaction. This
is a standard picture for nonbiological catalysis, and it also
has significant experimental support. A critical observation
is found using kinetic isotope effect methods. In this way,
one can probe the chemical structure of the transition state
in the catalytic event. Stable molecules can be designed that
share the electronic properties of the transition state (usually
identified by the electrostatic potential at the van der Waals
surface). Furthermore, these molecules make highly potent
inhibitors [5,6]. When substrate-like molecules that cannot
react to form products bind, often a far lower level of
inhibition is found. This result is said to be indicative of the
fact that the transition state is strongly bound. It has been
argued, however, that the electrostatic character of the
active site during the catalytic event is largely determined by
whatever charge stabilization is needed as the reaction
progresses. If an inhibitor is designed with the complement-
ary charges, it will bind strongly to the active site. However,
this does not imply that the method by which the enzyme
produced catalysis was transition state binding and con-
comitant release of energy [1].
A second approach, which might be viewed as the
converse of transition state stabilization, is ground state
destabilization. In this picture [7], the role of the enzyme is to
make the reactants less stable rather than making the
transition state more stable. Thus the energetic hill that must
be climbed with thermal activation is lowered. Energies are
all relative and so the end effect of this and the first
mechanism are the same; lowering the relative energy
difference between reactants and transition state. But it is
clear that this view presents a very different physical
mechanism. Recent calculations [8] seem to show that this
model may well be dominant for the most efficient enzyme
known, orotidine monophosphate decarboxylase.
A third concept that has been also suggested. In solution,
reactants are strongly solvated by water, the dominant
component of most living cells. When enzymes bind
reactants, they often exclude water, and this lowered
dielectric environment may be more conducive to reaction
[9–11]. This approach to catalysis tends to treat the catalytic
event much like an electron transfer reaction in solution.
The dominant description of electron transfer in solution is
Marcus’ theory [12], and this approach has also been used to
describe atom transfer [13]. The concept here is that the
main barrier to reaction is, in fact, reorganization of the
solvent as charged particles move, rather than the intrinsic
chemical barrier due to transformation of the substrate. It is
certainly true that such energy reorganization may be a
significant component in many cases, but probably does not
account for all catalysis in biological systems.
A fourth recent suggestion by Bruice [14,15] is that the
dominant role of an enzyme is to position substrates in such
a way that thermal fluctuations easily take them over the
barrier to reaction. The set of positions the enzyme
encourages the substrate to take are known as near attack
conformations(NACs). Here, while the enzyme might bind
strongly to a transition state structure, this binding energy is
not thought to be released specifically to speed the reaction.
The enzyme moulds the substrate so that it is on the edge of
reacting and forming products. Because the enzyme helps
the reactants to form the NAC, this view is philosophically a
bit closer to the ground state destabilization view. It is,
however, not a statistical energetic argument, but rather a
chemical structure argument.
A fifth possibility for the mode of action of enzymes is the
principle subject of this paper, that is, motions within the
protein itself actually speed the rate of a chemical reaction.
There is significant relation between this possibility and the
last view of catalysis described above, i.e. the creation of the
NAC. It must be stressed, however, that the current view is a
dynamic one. For this concept to be true, actual motions of
the protein must couple strongly to a reaction coordinate
and cause an increase in reaction rate. This is not simply
preparation of a reactive species, but rather dynamic
coupling. It is important to note that this is an entirely
different view of the method by which the enzyme
accomplishes rate acceleration. In this view, evolution has
created a protein structure that moves in such a way as to
lower a barrier and make it less wide. It must be emphasized
that this lowering of the barrier is not statistical lowering of
a potential of mean force through the release of binding
energy, but rather the use of highly directed energy (a
vibration) in a specific direction. Furthermore, this is not
simply the statistical preparation of reactive species as in the
NAC concept. Here, protein dynamics directly affect the
reaction coordinate potential. Although this effect can be
quite apparent for a tunneling system (the probability to
tunnel increases exponentially with a reduction of the width
of the tunneling barrier), it is equally important for systems
where the reaction proceeds through classical transfer,
because as the barrier is made narrower, it is also lowered.
In order to understand how directed protein motions may
cause catalysis, we need a theory of chemical reactions in a
condensed phase. Our group has developed theories over
the past 10 years, and this work, initially developed for
simple condensed phases, such as polar media, forms the
basis for our analysis. We now describe these theories in
some detail.
AN ENZYME AS A CONDENSED PHASE:
THEORETICAL FORMULATION FOR
THE STUDY OF CHEMICAL REACTION
There are two requirements to enable the study of a
chemical reaction in any system, be it as simple as a gas
phase collision, or as complex as that in an enzyme. First, a
potential energy for the interaction of all the atoms in the
system is needed. This includes the interactions of all atoms
having their chemical bonds changed, and those that do not.
The second requirement is for a method to solve the
dynamics of the equations of motion that allow one to
follow the progress of the reacting species in the presence of
the rest of the system from reactants to products. In this
work, we assume that we are able to obtain the first
requirement (the potential). In order to study the dynamics
on this potential, however, one needs to solve the Schro-
dinger equation for the entire collection of atoms. It is a
well-known fact that this is difficult for three or four atoms,
and so essentially impossible for the thousands of atoms in a
reaction catalyzed by an enzyme.
Various groups have taken a number of possible
approaches to solve this problem. One may assume that
quantum effects are minor, and use a purely classical
3104 D. Antoniou et al. (Eur. J. Biochem. 269)FEBS 2002

approach to solve the dynamics [16]. We are specifically
interested in studies of enzyme systems where quantum
mechanics plays a significant role, through tunneling of a
light particle, in the chemical step of the enzyme, and so the
classical approach will not be expected to yield valid results.
Another approach is to use a mixed quantum-classical
formulation in which a subset of the atoms is treated
quantum mechanically and the rest of the system is treated
purely classically. In recent years, this approach has become
popular with the pioneering work of such investigators as
Gao [8]. We have chosen a different approach, largely on
stylistic grounds. Rather than treating the full collection of
atoms as a mixture of quantum and classical objects
(something that is difficult to define rigorously), we have
developed approximate approaches to treat the entire
collection of atoms as a quantum mechanical entity. As
mentioned above, both approaches are approximate, but we
prefer to make the approximation uniform for the entire
system.
We have called our approach the Quantum Kramers
methodology [17,18]. Our ideas were motivated by the
following approximations developed for the study of the
classical mechanics of large, complex systems. It is known
that for a purely classical system [19,20], an accurate
approximation of the dynamics of a tagged degree of
freedom (for example a reaction coordinate) in a condensed
phase can be obtained through the use of a generalized
Langevin equation. The generalized Langevin equation is
given by Newtonian dynamics plus the effects of the
environment in the form of a memory friction and a random
force [21]. Thus, all the complex microscopic dynamics of all
degrees of freedom other than the reaction coordinate are
included only in a statistical treatment, and the reaction
coordinate plus environment is treated as a modified one-
dimensional system. What allows a realistic simulation of
complex systems is that the statistics of the environment can
in fact be calculated from a formal prescription. This
prescription is given by the fluctuation-dissipation the-
orem, which yields the relationship between friction and
random force. In particular, this theory enables us to
calculate the memory friction from a relatively short-time
classical simulation of the reaction coordinate. The Quan-
tum Kramers approach, in turn, is dependent on an
observation of Zwanzig [22,23]; if an interaction potential
for a condensed phase system satisfies a fairly broad set of
mathematical criteria, the dynamics of the reaction coordi-
nate as described by the generalized Langevin equation can
be rigorously equated to a microscopic Hamiltonian in
which the reaction coordinate is coupled to an infinite set of
Harmonic Oscillators via simple bilinear coupling:
H¼P2
s
2ms
þVoþX
k
P2
k
2mk
þ1
2mkx2
kqkcks
mkx2
k
2
ð1Þ
The first two terms in this Hamiltonian represent the kinetic
and potential energy of the reaction coordinate, and the last
set of terms similarly represent the kinetic and potential
energy for an environmental bath. Here, srepresents some
coordinate that measures progress of the reaction (for
example, in alcohol dehydrogenase where the chemical
step is transfer of a hydride, smight be chosen to represent
the relative position of the hydride from the alcohol to the
NAD cofactor.) c
k
is the strength of the coupling of the
environmental mode to the reaction coordinate, and m
k
and x
k
give the effective mass and frequency, respectively,
of the environmental bath mode. A discrete spectral density
gives the distribution of bath modes in the harmonic
environment:
JðxÞ¼p
2X
k
c2
k
mkxk
dðxxkÞdðxþxkÞ
½
ð2Þ
Here d(x)x
k
)istheDirac deltafunction, so the spectral
density is simply a collection of spikes, located at the
frequency positions of the environmental modes, weighted
by the strength of the coupling of these modes to the
reaction coordinate. Note that this infinite collection of
oscillators is purely fictitious; they are chosen to reproduce
the overall physical properties of the system, but do not
necessarily represent specific physical motions of the atoms
in the system. It would seem that we have not made a huge
amount of progress; we began with a many-dimensional
system (classical) and found out that it could be accurately
approximated by a one-dimensional system in a frictional
environment (the generalized Langevin equation.) We have
now recreated a many-dimensional system (the Zwanzig
Hamiltonian). The reason we have done this is twofold.
First, there is no true quantum mechanical analogue of
friction, and so there really is no way to use the generalized
Langevin approach for a quantum system, such as we
would like to do for an enzyme. Second, the new quantum
Hamiltonian given in Eqn (1) is much simpler than the
Hamiltonian for the full enzymatic system. Harmonic
oscillators are a problem that can easily be solved by
quantum mechanics. Thus, the prescription is, given a
potential for the enzymatic reaction, we model the exact
problem using Zwanzig Hamiltonian, as in Eqn (1), with
the distribution of harmonic modes given by the spectral
density in Eqn (2), and found through a simple classical
computation of the frictional force on the reaction coordi-
nate. Then, using methods to compute quantum dynamics
developed in our group [24–29], quantities such as rates or
kinetic isotope effects may be computed. Thus, the quantum
Kramers method, developed in our group, consists of the
following ingredients. Given a potential for the enzymatic
reaction, we model the exact problem using Zwanzig’s
Hamiltonian, as in Eqn (1), with the distribution of
harmonic modes given by the spectral density in Eqn (2).
The spectral density is obtained through a molecular
dynamicssimulation of the classical system. Then, using
methods developed in our group to carry out the quantum
dynamics, quantities such as rates or kinetic isotope effects
may be computed.
This approach enables us to model a variety of condensed
phase chemical reactions with essentially experimental
accuracy [30]. There are deeper connections between this
approach and another popular method of dynamics com-
putation in complex systems. We have shown [30] that this
collection of bilinearly-coupled oscillators is in fact a
microscopic version of the popular Marcus theory for
charged particle transfer [12,13]. The bilinear coupling of the
bath of oscillators is the simplest form of a class of couplings
that may be termed antisymmetric because of the mathe-
matical property of the functional form of the coupling on
reflection about the origin. This property has deeper
implications than the mathematical nature of the symmetry
FEBS 2002 Barrier passage and protein dynamics (Eur. J. Biochem. 269) 3105
properties. Antisymmetric couplings, when coupled to a
double-well-like potential energy profile, are able to instan-
taneously change the level of well depths, but do nothing to
the position of well minima. This modulation in the position
of minima is exactly what the environment is envisaged to
do within the Marcus theory paradigm. As we have shown
[30], the minima of the total potential in Eqn (1) will occur,
for a two-dimensional version of this potential, when the q
degree of freedom is exactly equal and opposite in sign to
cs
mx2, and the minimum of the potential energy profile along
the reaction coordinate is unaffected by this coupling.
Within Marcus’ theory, which is a deep tunneling theory,
transfer of the charged particle occurs at the value of the
bath coordinates that cause the total potential to become
symmetrized. Thus, if the bare reaction coordinate potential
is symmetric, then the total potential is symmetrized at the
position of the bath plus couplingminimum. When this
configuration is achieved, the particle tunnels; the activation
energy for the reaction is largely the energy to bring the bath
into this favorable tunneling configuration.
While Marcus’ theory and our microscopic quantum
Kramers theory are highly successful in many cases, in other
cases, it is not possible to reproduce experimental results
using such an approach. The reason for this is that the
antisymmetric coupling contained within the Zwanzig
Hamiltonian does not physically represent all possible
important motions in a complex reacting system. In fact,
such a reality was pointed out some time ago in seminal
work of the Hynes group [31]. In some of our earlier work
on hydrogen transfer in enzymatic systems, we were able to
show that one could reasonably fit experimental kinetic data
in such enzymatic systems with phenomenological applica-
tion of the Hynes theories [32]. We became interested in a
microscopic study of such systems in the examination of
nonbiological proton transfer reactions, i.e. organic acid
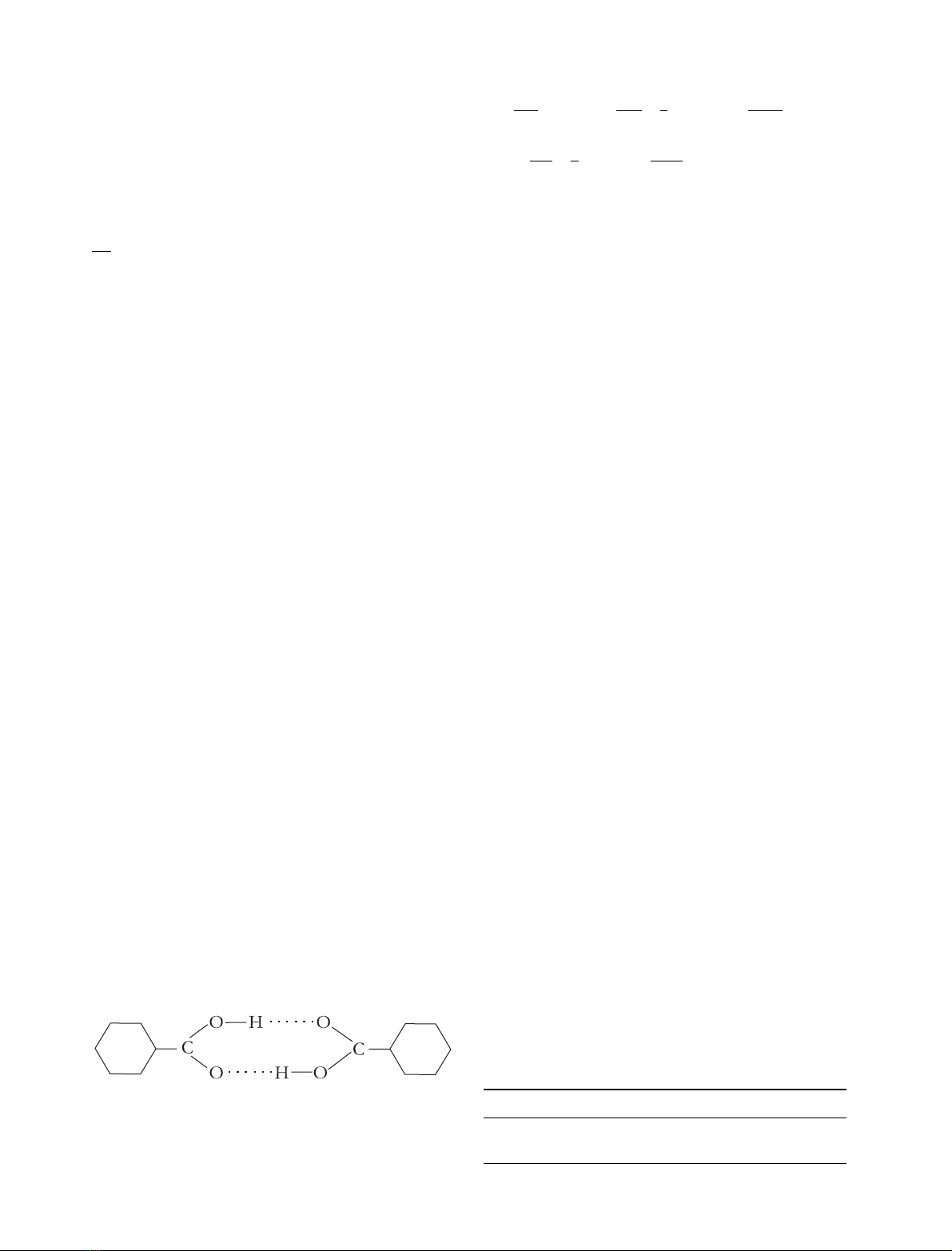
crystals. The simplest example is a carboxylic acid dimer,
showninFig.1.Suchsystemshadbeenstudiedformany
years [33–37], and they presented what seemed to be a
chemical physics conundrum. While quantum chemistry
computations seemed to show that the intrinsic barrier to
proton transfer in these systems was reasonably high, and
low experimental activation energies seemed to indicate a
significant involvement of quantum tunneling in the proton
transfer mechanism, careful measurements of kinetic iso-
tope effects showed kinetics indicative of classical transfer.
In order to study such systems, a rigorous theory, which
allowed inclusion of symmetrically coupled vibrations, in
addition to an environmental bath of antisymmetrically
coupled oscillators, was needed. Mathematically, the simp-
lest transformation of the Hamiltonian in Eqn (1) is given
by:
H¼P2
S
2ms
þVoþX
k
P2
k
2mk
þ1
2mkx2
kqkcks
mkx2
k
2
þP2
Q
2Mþ1
2mX2QCs2
MX2
ð3Þ
Note that in this case, the oscillator that is symmetrically
coupled, represented by the last term in Eqn (3), is in fact a
physical oscillation of the environment.
We were able to develop a theory [38] of reactions
mathematically represented by the Hamiltonian in Eqn (3),
and using this method and experimentally available param-
eters for the benzoic acid proton transfer potential, we were
able to reproduce experimental kinetics as long as we
included a symmetrically coupled vibration [39]. The results
are shown in Table 1 below. The two-dimensional activa-
tion energies refer to a two-dimensional system comprised
of the reaction coordinate and a symmetrically coupled
vibration. The reaction coordinate is also coupled to an
infinite environment as described above.
In this case, the symmetric motion has a clear physical
origin: the symmetric motion of the carbonyl and hydroxyl
oxygen atoms toward each other. Kinetic isotope effects in
this system are modest, even though the vast majority of the
proton transfer occurs via quantum tunneling. The end
result of this study is that symmetrically coupled vibrations
can significantly enhance rates of light particle transfer, and
also significantly mask kinetic isotope signatures of tunnel-
ing. A physical origin for this masking of the kinetic isotope
effect may be understood from a comparison of the two-
dimensional problem comprised of a reaction coordinate
coupled symmetrically and antisymmetrically to a vibration.
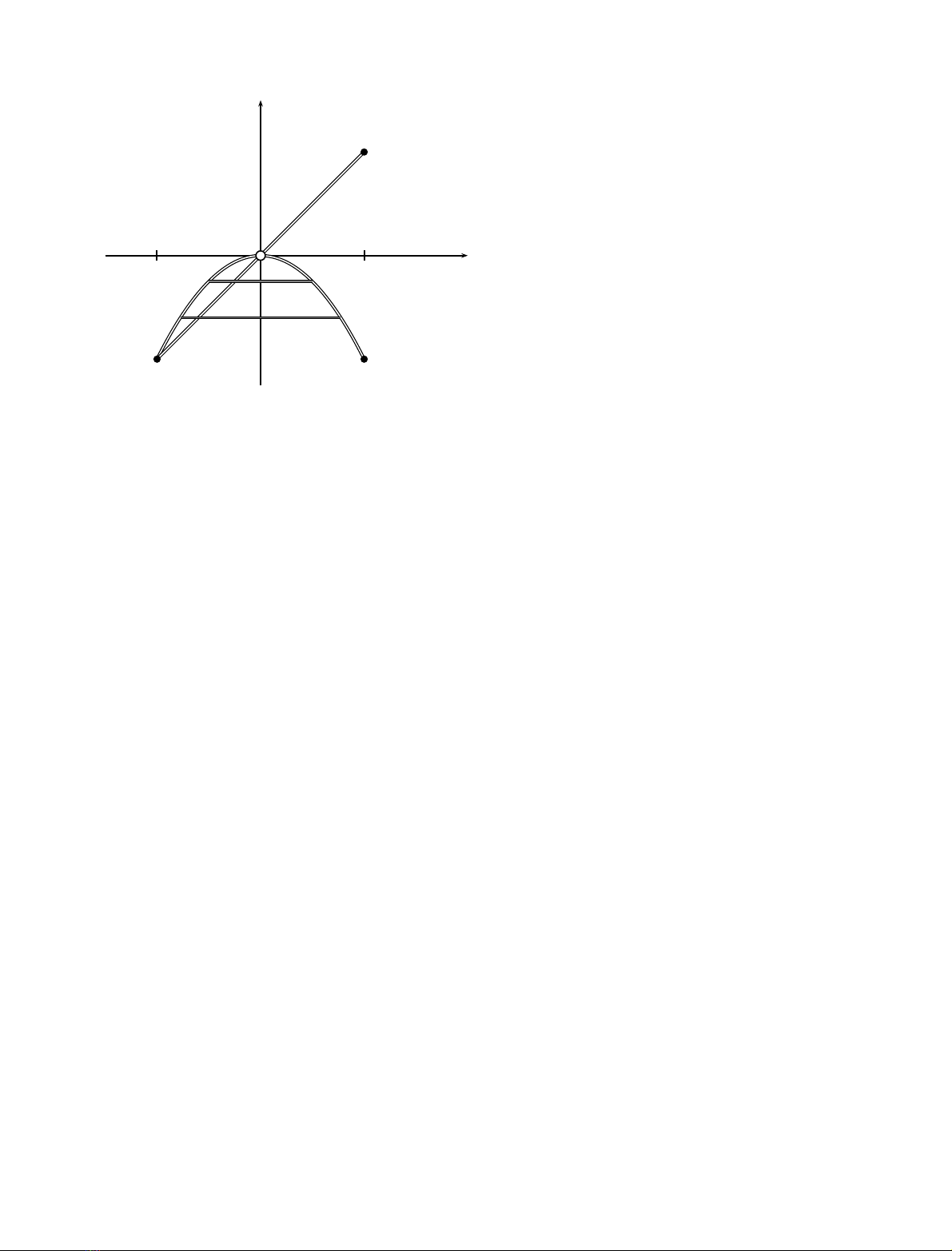
As Fig. 2 shows, antisymmetric coupling causes the minima
(the reactants and products) to lie on a line; the minimum
energy path, which passes through the transition state. In
contrast, symmetric coupling causes the reactants and
products to be moved from the reaction coordinate axis in
such a way that a straight line connection of reactant and
products would pass no where near the transition state.
This, in turn, results in the gas phase physical chemistry
phenomenon known as corner cutting [40–42]. Physically,
the quantity to be minimized along any path from reactant
to products is the action. This is an integral of the energy,
and so loosely speaking, it is a product of distance and depth
under the barrier that must be minimized to find an
approximation to the tunneling path. The action also
includes the mass of the particle being transferred, and so in
the symmetric coupling case, a proton will actually follow a
very different physical path from reactants to products in a
reaction than a deuteron. (Not just in the trivial sense that
one tunnels more than another). It is this following of a
different physical path, even when tunneling dominates,
Fig. 1. A benzoic acid dimer. Thereactioncoordinateinthiscaseisthe
symmetric transfer of the hydroxyl protons to the carbonyl oxygen.
The promoting vibration is the symmetric motion of the oxygens
toward each other.
Table 1. Activation energies for H and D transfer in benzoic acid
crystals at T ¼300 K. Three values are shown: the activation energies
calculated using a one- and two-dimensional Kramers problem and the
experimental values. The values of energies are in kcalÆmol
)1
.
E
1d
E
2d
Experiment
H 3.39 1.51 1.44 kcalÆmol
)1
D 5.21 3.14 3.01 kcalÆmol
)1
3106 D. Antoniou et al. (Eur. J. Biochem. 269)FEBS 2002
that causes the kinetic isotope effects to be masked. It was
this low level of primary kinetic isotope effect that suggested
a similarity between the proton transfer mechanism in the
organic acid crystal and that of enzymatic reactions. While
coupled motions of nearby atoms in enzymatic reactions
have been used to explain anomalous kinetic isotope effects
[43], these were studies in a classical picture with semiclas-
sical tunneling added (the Bell correction; [44]) and they
could not be used to account for enzymatic reactions in a
deep tunneling regime.
Klinman and coworkers have helped pioneer the study of
tunneling in enzymatic reactions. One focus of their work
has been the alcohol dehydrogenase family of enzymes.
Alcohol dehydrogenases are NAD
+
-dependent enzymes
that oxidize a wide variety of alcohols to the corresponding
aldehydes. After successive binding of the alcohol and
cofactor, the first step is generally accepted to be complex-
ation of the alcohol to one of the two bound Zinc ions [45].
This complexation lowers the pK
a
of the alcohol proton and
causes the formation of the alcoholate. The chemical step is
then transfer of a hydride from the alkoxide to the NAD
+
cofactor. They [46] have found a remarkable effect on the
kinetics of yeast alcohol dehydrogenase (a mesophile) and a
related enzyme from Bacillus stereothermophilus, a thermo-
phile. A variety of kinetic studies from this group have
found that the mesophile [47] and many related dehydro-
genases [48–51] show signs of significant contributions of
quantum tunneling in the rate-determining step of hydride
transfer. Remarkably, their kinetic data seem to show that
the thermophilic enzyme actually exhibits less signs of
tunneling at lower temperatures. Recent data of Kohen &
Klinman [52] also show, via isotope exchange experiments,
that the thermophile is significantly less flexible at mesophi-
lic temperatures, as in the results of Petsko et al. [53], who
conducted studies of 3-isopropylmalate dehydrogenase
from the thermophilic bacteria Thermus thermophilus.These
data have been interpreted in terms of models similar to
those we have described above, in which a specific type of
protein motion strongly promotes quantum tunneling; thus,
at lower temperatures, when the thermophile has this
motion significantly reduced, the tunneling component of
reaction is hypothesized to go down even though one would
normally expect tunneling to go up as temperature goes
down. Additionally, the Klinman group has investigated the
catalytic properties of various mutants of horse liver alcohol
dehydrogenase (HLADH). HLADH in the wild-type has a
slightly less advantageous system to study than yeast
alcohol dehydrogenase, because the chemistry is not the
rate determining step in catalysis for this enzyme. Two
specific mutations have been identified, Val203 fiAla and
Phe93 fiTrp, which significantly affect enzyme kinetics.
Both residues are located at the active site; the valine
impinges directly on the face of the NAD
+
cofactor distal to
the substrate alcohol. Modification of this residue to the
smaller alanine significantly lowers both the catalytic
efficiency of the enzyme, as compared to the wild-type,
and also significantly lowers indicators of hydrogen tunnel-
ing [54]. Phe93 is a residue in the alcohol binding pocket.
Replacement with the larger tryptophan makes it harder for
the substrate to bind, but does not lower the indicators of
tunneling [55]. Bruice’s recent molecular dynamics calcula-
tions [56] produce results consistent with the concept that
mutation of the valine changes protein dynamics, and it is
this alteration, missing in the mutation at position 93, which
in turn changes tunneling dynamics. (We note the recent
experimental results from Klinman’s group [57] in which no
decrease in tunneling is seen as the temperature is raised.)
A final set of enzymes now thought to exhibit dynamic
protein control of tunneling hydrogen transfer is that in the
amine dehydrogenase family. Scrutton and coworkers have
extensively studied these enzymes [58]. Though similarly
named and having a similar end effect as the alcohol
dehydrogenases, they employ radically different chemistry.
These enzymes catalyze the oxidative deamination of
primary amines to aldehydes and free ammonia. In this
case, however, rather than a chemical step of hydride
transfer, the rate determining chemical step is proton
transfer; and in fact these enzymes catalyze a coupled
electron proton transfer reaction. Electrons are coupled to
some cofactor, for example, in the case of aromatic amine
dehydrogenase, the cofactor is tryptophan-tryptophyl qui-
none. Kinetic studies have shown that methylamine dehy-
drogenase exhibits not only relatively large primary kinetic
isotope effects (unlike the alcohol dehydrogenases), but also
very strong temperature dependence in the measured
activation energy. This experimental data has been inter-
preted as showing that the enzyme works via a promoting
vibration [59], as we have suggested for bovine serum amine
oxidase [32], and for various forms of HLADH [60]. Here,
the primary kinetic isotope effect is 17, rather than 3 or 4.
s
q
s0+s
0
A,S S
A
Fig. 2. This diagram shows the location of stable minima in two-
dimensional systems. In one case a vibrational mode is symmetrically
coupled to the reaction coordinate, and in the other, antisymmetrically
coupled. The figure represents how antisymmetrically and symmetri-
cally coupled vibrations effect position of stable minima – that is
reactant and product wells – in modulating the one dimensional double
well potential (before coupling along the xaxis). The xaxis, s,repre-
sents the reaction coordinate, and qthe coupled vibration. The points
on the figure labeled S and A are the positions of the well minimal in
the two dimensional system with symmetric and antisymmetric coup-
ling, respectively. An antisymmetrically coupled vibration displaces
those minima along a straight line, so that the shortest distance
between the reactant and product wells passes through the transition
state. In contradistinction, a symmetrically coupled vibration, allows
for the possibility of corner cuttingunder the barrier. For example, a
proton and a deuteron will follow different paths under the barrier.
FEBS 2002 Barrier passage and protein dynamics (Eur. J. Biochem. 269) 3107

























