
Identification of domains on the extrinsic 23 kDa protein possibly
involved in electrostatic interaction with the extrinsic 33 kDa protein
in spinach photosystem II
Akihiko Tohri
1,2
, Naoshi Dohmae
3
, Takehiro Suzuki
1
, Hisataka Ohta
1,4
, Yasunori Inoue
2,4
and Isao Enami
1
1
Department of Biology, Faculty of Science, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo, Japan;
2
Department of Applied Biological Science, Faculty of Science and Technology, Tokyo University of Science, Yamazaki,
Noda, Chiba, Japan;
3
Division of Biochemical Characterization, the Institute of Physical and Chemical Research (RIKEN),
Hirosawa, Wako, Saitama, Japan;
4
Tissue Engineering Research Center, Tokyo University of Science, Yamazaki, Noda, Chiba, Japan
To elucidate the domains on the extrinsic 23 kDa protein
involved in electrostatic interaction with the extrinsic 33 kDa
protein in spinach photosystem II, we modified amino or
carboxyl groups of the 23 kDa protein to uncharged methyl
ester groups with N-succinimidyl propionate or glycine
methyl ester in the presence of a water-soluble carbodi-
imide, respectively. The N-succinimidyl propionate-modified
23 kDa protein did not bind to the 33 kDa protein associ-
ated with PSII membranes, whereas the glycine methyl ester-
modified 23 kDa protein completely bound. This indicates
that positive charges on the 23 kDa protein are important for
electrostatic interaction with the 33 kDa protein associated
with the PSII membranes. Mapping of the N-succinimidyl
propionate-modified sites of the 23 kDa protein was per-
formed using Staphylococcus V8 protease digestion of the
modified protein followed by determination of the mass of
the resultant peptide fragments with MALDI-TOF MS. The
results showed that six domains (Lys11–Lys14, Lys27–
Lys38, Lys40, Lys90–Lys96, Lys143–Lys152, Lys166–
Lys174) were modified with N-succinimidyl propionate. In
these domains, Lys11, Lys13, Lys33, Lys38, Lys143, Lys166,
Lys170 and Lys174 were wholly conserved in the 23 kDa
protein from 12 species of higher plants. These positively
charged lysyl residues on the 23 kDa protein may be involved
in electrostatic interactions with the negatively charged
carboxyl groups on the 33 kDa protein, the latter has been
suggested to be important for the 23 kDa binding [Bricker,
T.M. & Frankel, L.K. (2003) Biochemistry 42, 2056–2061].
Keywords: extrinsic 23 kDa protein; extrinsic 33 kDa pro-
tein; electrostatic interaction; chemical modification; oxygen
evolution.
Photosystem II (PSII) catalyzes the light-driven oxidation
of water with concomitant reduction of plastoquinone to
plastoquinol. This multisubunit protein-pigment complex
contains a number of intrinsic proteins and 3–4 extrinsic
proteins associated with the lumenal side of PS II. The three
extrinsic proteins of 33, 23 and 17 kDa associate with higher
plant and green algal PSII [1]. Their binding properties,
however, are different between higher plant and green algal
PSII. In higher plant PSII, the 33 kDa protein associates
directly with PSII, but the 23 kDa protein cannot directly
bind to PSII and associates with PSII only through its
interaction with the 33 kDa protein, and the 17 kDa protein
functionally associates with PSII only through its inter-
action with both the 33 and 23 kDa proteins [2]. The 23 and
17 kDa proteins are easily released from higher plant PSII
by washing with 1
M
NaCl, indicating that the 23 kDa
protein electrostatically binds to the 33 kDa protein [3], and
the 17 kDa protein interacts electrostatically with both the
33 and 23 kDa proteins. In contrast, the green algal 23 and
17 kDa proteins can bind directly to PSII independent of
the presence or absence of other extrinsic proteins [4]. On
the other hand, cyanobacterial PSII contains three extrinsic
proteins of 33 and 12 kDa, and cytochrome c550 [5],
whereas, red algal PSII contains four extrinsic proteins of
33, 20 and 12 kDa, and cytochrome c550 [6,7].
The extrinsic proteins play important roles for maximal
rates of oxygen evolution under physiological ionic condi-
tions [1]. The 33 kDa protein is needed to maintain the
functional conformation of the Mn cluster [8,9]. Shutova
et al. found that titration of the 33 kDa protein against pH
in solution exhibited a striking hysteresis [10], and proposed
that the protein is not only required for stabilizing the
Mn-cluster but also important for proton transport to occur
appropriately, accompanying oxygen evolution [11]. The
functions of the 23 and 17 kDa proteins are closely related
with the unique requirement of Ca
2+
and Cl
–
for oxygen
evolution; the 23 kDa protein mitigates the demand for
Ca
2+
while the 17 kDa protein does for Cl
–
[8,12–14].
Correspondence to I. Enami, Department of Biology, Faculty of
Science, Tokyo University of Science, Kagurazaka 1-3, Shinjuku-ku,
Tokyo 162-8601, Japan. Tel.: + 81 4 7124 1501 (ext. 5022),
E-mail: enami@rs.noda.tus.ac.jp
Abbreviations: CBB, Coomasie brilliant blue; Chl, chlorophyll; CHC,
a-cyano-4-hydroxycinnamic acid; DHB, 2,5-dihydroxybenzoic acid;
EDC, 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide; GME,
glycine methyl ester; MBT, 2-mercaptobenzothiazole;
NHS, N-hydroxysuccinimido; NSP, N-succinimidyl propionate;
PSII, photosystem II.
(Received 28 October 2003, revised 9 January 2004,
accepted 16 January 2004)
Eur. J. Biochem. 271, 962–971 (2004) ÓFEBS 2004 doi:10.1111/j.1432-1033.2004.03998.x

The extrinsic proteins of 12 kDa and cytochrome c550
in cyanobacterial and red algal PSIIs have a similar func-
tion to that of the 23 and 17 kDa proteins in higher
plant PSII [7,15–17].
Recently, Zouni et al. [18] and Kamiya and Shen [19]
published the crystal structural analysis of thermophilic
cyanobacterial PSII. These studies have provided important
insights into the organization of numerous subunits of
cyanobacterial PSII. The 33 kDa protein and cytochrome
c550 appear to interact with the large extrinsic loop E of
CP47 and with the large extrinsic loop E of CP43, respect-
ively. The 12 kDa protein appears to interact with both the
33 kDa protein and cytochrome c550 [19]. These studies
provided, however, few insights into the structural organ-
ization of the 23 and 17 kDa proteins in higher plant PSII.
Three-dimensional crystals from higher plant PSII uniformly
diffract poorly [20] and two-dimensional crystals examined
by electron diffraction have been performed at low
resolutions with PSII from higher plants devoid of an
oxygen-evolving complex [21,22]. In cross-reconstitution
experiments, the 23 and 17 kDa proteins bound to cyano-
bacterial and red algal PSII only through non-specific
interactions [16]. The CaCl
2
-washed spinach PS II mem-
branes which had been reconstituted with either cyanobac-
terial or red algal 33 kDa protein, could only partially rebind
spinach 23 kDa protein but could not bind spinach 17 kDa
protein [23]. These data indicate that there are structural
determinants present on the spinach 33 kDa protein that are
required for the efficient binding of the 23 and 17 kDa
proteins and that are absent in cyanobacterial and red algal
proteins [24].
The organization among the three extrinsic proteins
in spinach PSII has been examined by cross-linking
experiments. Cross-linking experiments performed with
homobifunctional cross-linkers (6–14 A
˚span) indicated
that the 33 kDa protein is within a distance of 11 A
˚of
the 23 kDa protein and that the 23 kDa protein is within
11 A
˚of the 17 kDa protein [25]. This indicates that these
three extrinsic proteins must be in close proximity. Cross-
linking experiments also showed that the 33 kDa protein is
associated with or in close proximity to CP47 [25–28],
D1 and D2 [29], a large subunit of cytochrome b559 [30]
and PsbI [30]. The 33 kDa protein was shown to be also
associated with CP43 by comparing the peptide mappings
of the trypsin-digested products of NaCl-washed and
CaCl
2
-washed PSII membranes [31]. Thus, the 33 kDa
protein is associated with or in close proximity to essentially
all of the major intrinsic proteins in higher plant PSII.
Chemical modification is a useful method to elucidate
which positive or negative charges on the extrinsic proteins
are responsible for electrostatic interaction with the other
extrinsic proteins and/or the intrinsic proteins [32,33]. We
have reported that the N-succinimidyl propionate (NSP)-
modified 33 kDa protein, of which the positively charged
amino groups are modified to uncharged methyl ester groups
[33], cannot rebind to spinach PSII, whereas the glycine
methyl ester (GME)-modified protein, of which the negat-
ively charged carboxyl groups are modified to uncharged
methyl ester groups [32], can rebind and reactivate the
oxygen evolution [34]. These results indicate that positive
charges on the 33 kDa protein are important for its
electrostatic interaction with PSII intrinsic proteins, whereas
negative charges on the protein do not contribute to such
interaction. The domains of the 33 kDa protein possibly
involved in electrostatic interaction with PSII intrinsic
proteins were also determined to be Lys4, Lys20, Lys66–
Lys76, Lys101, Lys105, Lys130, Lys159, Lys186 and
Lys230–Lys236 by a combination of V8 protease digestion
and MALDI-TOF MS of NSP or 2,4,6-trinitrobenzene
sulfonic acid-modified 33 kDa protein [34], or NHS-biotin
modified one [35]. Furthermore, we showed that a similar
number of carboxyl groups on the 33 kDa protein were
modified with GME in both the protein in solution and
bound to PSII [34]. This suggests that most of the carboxyl
groups on the 33 kDa protein are not located in regions
interacting with PSII intrinsic proteins and exposed to the
lumenal side of PSII. Thus, we hypothesized that negative
charges of carboxyl groups on the 33 kDa protein may be
involved in electrostatic interaction with the 23 and 17 kDa
proteins. In fact, Bricker and Frankel [24] showed recently,
that spinach PS II membranes reconstituted with the 33 kDa
protein, on which the negatively charged carboxyl groups
were modified with GME, was defective in its ability to bind
the 23 kDa protein of PSII. They hypothesized that the
domains on the 33 kDa protein possibly involved in
electrostatic interaction with the 23 kDa protein are Glu1,
Glu32, Glu139 and/or Glu187, which are wholly conserved
in higher plants but which are poorly conserved in cyano-
bacteria. These facts in turn suggest that positive charges on
the 23 kDa protein may be responsible for the electrostatic
interaction with these negative charges on the 33 kDa
protein.
The binding domains of the 23 kDa protein, however,
remain obscure. Recently, Ifuku and Sato [36] reported that
the binding affinity of a recombinant mutant of the 23 kDa
protein, of which N-terminal 19 residues were truncated,
were apparently weaker than that of the native 23 kDa
protein, and the mutant protein completely lacked the
ability to retain Ca
2+
for oxygen evolution. This suggests
that the N-terminal region of the 23 kDa protein is
important for its binding with the 33 kDa protein.
In the present study, the domains on the 23 kDa protein
possibly involved in electrostatic interaction with the 33 kDa
protein associated with PSII membranes were examined
by chemical modification method. The results showed that
positive charges on the 23 kDa protein are indeed important
for its interaction with the 33 kDa protein, and we have
determined the domains of positive charges on the 23 kDa
protein that are possibly involved in the interaction.
Materials and methods
Preparations
Oxygen-evolving PSII membranes were prepared from
spinach chloroplasts with Triton X-100 as described in
Berthold et al. [37], with slight modifications [28]. The
isolated PSII membranes were suspended in medium A
(40 m
M
Mes/NaOH,pH6.5;0.4
M
sucrose; 10 m
M
NaCl
and 5 m
M
MgCl
2
, and stored in liquid nitrogen until used.
The extrinsic 33 and 23 kDa proteins were extracted from
the PSII membranes by 1
M
CaCl
2
treatment, incubated
with 1
M
CaCl
2
for 3 h in the dark to suppress the activity
of copurified protease, dialyzed against 5 m
M
Mes/NaOH,
ÓFEBS 2004 Interaction between the 23 and 33 kDa proteins (Eur. J. Biochem. 271) 963

pH 6.5 and further against 20 m
M
phosphate buffer, pH 6.5
and then purified by column chromatography with a
DEAE-Sepharose CL-6B column (Pharmacia Biotech Inc.,
NJ, USA) [16,38]. The concentrations of the 33 and 23 kDa
proteins were determined using an extinction coefficient of
16 m
M
)1
Æcm
)1
at 276 nm [39] and 26 m
M
)1
Æcm
)1
at 277 nm
[38], respectively.
Chemical modification
For modification of amino groups of lysyl residues and the
free amino terminus of the 23 kDa protein, the purified
protein (30 l
M
) was incubated in a reaction mixture
containing 20 m
M
phosphate buffer, pH 6.5 and
0.5–6.0 m
M
NSP at 25 °C for 90 min. The reaction mixtures
were dialyzed against 10 m
M
Mes/NaOH, pH 6.5 to
remove unreacted NSP. Chemical modification of carboxyl
groups on the purified 23 kDa protein was performed in
100 m
M
GME, pH 6.2 containing 30 l
M
of the 23 kDa
protein and 2 m
M
1-ethyl-3-(3-(dimethylamino)propyl) car-
bodiimide (EDC) at 25 °C for 12 h. The reaction mixture
was dialyzed against 1
M
NaCl and 20 m
M
phosphate
buffer, pH 6.5 to remove unreacted and electrostatically
attached reagents, and then against 10 m
M
Mes/NaOH,
pH 6.5. NSP was purchased from Wako Pure Chemicals
(Tokyo, Japan), and GME and EDC were purchased from
Nacalai Tesque Chemicals (Tokyo, Japan).
Reconstitution and electrophoresis
For reconstitution, PS II membranes were washed with
2.6
M
Urea, 0.2
M
NaCl in the dark to remove the three
extrinsic proteins of 33, 23 and 17 kDa [8]. The resultant
PSII membranes were incubated with the 33 kDa protein
and with either the unmodified or modified 23 kDa protein
at a protein-Chl ratio of 0.6 (w/w), in medium A at 0 °Cfor
30 min in the dark at a Chl concentration of 0.5 mg mL
)1
.
The reconstituted PSII membranes were collected by
centrifugation at 35 000 gfor 10 min and then washed
once with and resuspended in medium A. The reconstituted
PSII membranes were again treated with 2.6
M
urea, 0.2
M
NaCl in the dark for 30 min and the centrifuged super-
natants were applied on SDS/PAGE to estimate the
amounts of the 33 kDa and 23 kDa proteins rebound by
the reconstitution.
SDS/PAGE was performed with a gradient gel of 16–
22% acrylamide containing 7.5 urea [40]. Samples were
solubilized with 5% lithium lauryl sulfate and 75 m
M
dithiothreitol. The amounts of rebound 23 kDa protein
were determined from the integrated optical densities of the
23 kDa bands using the program
NIH IMAGE
(National
Institutes of Health, USA) after the SDS/PAGE was
scanned using a CanoScan N656U (Canon, Tokyo).
Isoelectric focusing was performed using a 5.5% poly-
acrylamide containing homogenous gel covering a pH range
of 3.5–10.0 or 4.0–6.0 using 5% (v/v) ampholine (Amer-
sham Pharmacia Biotech AB, Sweden). Proteins were
stained with 0.048% CBB in 30% methanol and 10%
acetic acid.
Oxygen evolution was measured with a Clark-type
oxygen electrode in 40 m
M
Mes/NaOH, pH 6.5 and 0.4
M
sucrose (medium B) at 25 °C in the absence and presence of
10 m
M
NaCl or 5 m
M
CaCl
2
, with 0.4 m
M
phenyl-
p-benzoquinone as the electron acceptor.
Chl concentration was determined by the method of
Porra et al. [41].
Protease digestion
The 23 kDa protein (3 nmol) modified with 0.5 or 4 m
M
NSP was dried and solubilized in 10 lLof1
M
Tris/HCl,
pH 8.5, 8
M
guanidine/HCl, 1 m
M
EDTA and 1% dithio-
threitol, and incubated at 37 °C for 2 h to denature the
23 kDa protein. Then, 5 lL of 5% iodoacetamide was added
and incubated at 37 °C for 30 min to block SH groups. The
reaction mixtures were added to a final concentration of 10%
of cold trichloroacetic acid and centrifuged, and the resulting
precipitates were washed twice with acetone. The final
precipitates were dried and resolubilized in 20 lLof0.1
M
ammonium bicarbonate. After 1 lgofStaphylococcus V8
protease (ICN Biomedicals, OH, USA) was added, the
23 kDa protein was digested at 37 °C, overnight and then
desalted by Ziptipl-C18 (Millipore, MA, USA).
Mass spectroscopic analysis
The protease-digested protein was applied directly to a
MALDI-TOF MS (Reflex; Bruker Daltonics, MA, USA),
with a matrix of a-cyano-4-hydroxycinnamic acid (CHC),
2-mercaptobenzothiazole (MBT) or 2,5-dihydroxybenzoic
acid (DHB). The mass of each measured peptide fragment
was assigned to the known 23 kDa protein sequence.
Results
As described above, Bricker and Frankel [24] showed that
negatively charged carboxyl groups on the extrinsic 33 kDa
protein are important for electrostatic interaction with the
extrinsic 23 kDa protein. This suggests that positive charges
on the 23 kDa protein may electrostatically interact with the
negative charges on the 33 kDa protein. To confirm this, we
modified positively charged amino groups on the 23 kDa
protein to uncharged methyl ester groups with NSP.
Figure 1A shows the isoelectric focusing of the NSP-
modified 23 kDa protein. The pI value shifted toward
acidic pH with increasing NSP concentration. For exam-
ple, the pI value downshifted from 6.8 (unmodified
protein, lane 1) to 4.8–5.5 (0.5 m
M
NSP-modified protein,
lane 2) and 4.3–4.8 (4 m
M
NSP-modified protein, lane 5).
These changes were estimated to result from modification
of 1–5 amino groups in 0.5 m
M
NSP-modified protein
and 5–10 amino groups in 4 m
M
NSP-modified protein to
uncharged groups, as calculated using a computer pI/Mr
tool [42]. It should be noted here that the band of the
modified protein appeared much broader than the
unmodified protein upon isoelectric focusing, implying
that the resulting protein products may be composed of
proteins with different numbers of amino residues modi-
fied. This is similar to the results obtained by modification
of the 33 kDa protein with NHS-biotin [35], NSP and
2,4,6-trinitrobenzen sulfonic acid [34], or GME [24].
In order to determine whether elimination of surface
positive charges affected binding of the 23 kDa protein,
the ability of the NSP-modified protein to rebind with the
964 A. Tohri et al. (Eur. J. Biochem. 271)ÓFEBS 2004
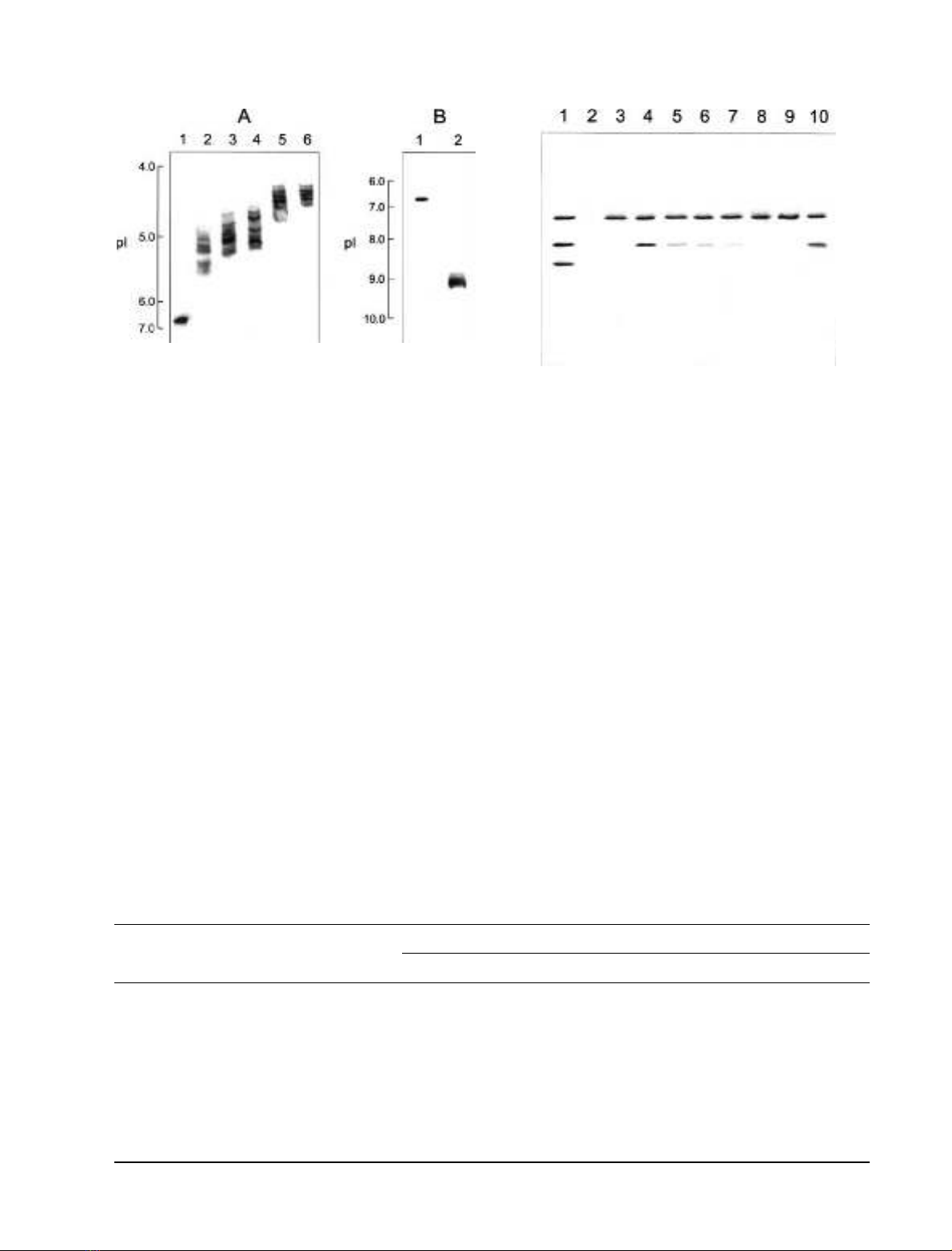
33 kDa protein associated with PSII membranes was
examined. Urea/NaCl-washed PSII membranes in which
the three extrinsic proteins of 33, 23 and 17 kDa had been
removed, were reconstituted with the unmodified and NSP-
modified 23 kDa protein together with the 33 kDa protein.
The reconstituted PSII membranes were again treated with
2.6
M
urea plus 0.2
M
NaCl, and the supernatants after
centrifugation were analyzed by SDS/PAGE to determine
the amounts of the 33 and 23 kDa proteins rebound. As
shown in Fig. 2, the native 33 and 23 kDa proteins
completely rebound to urea/NaCl-washed PSII membranes
(lane 4), whereas the binding abilities of NSP-modified
23 kDa protein decreased with increasing NSP concentra-
tion (lanes 5–9) and this ability was completely lost with
NSP treatments above 4 m
M
(lanes 8 and 9). This suggests
that positive charges on the 23 kDa protein are important
for electrostatic interaction with the 33 kDa protein.
Table 1 shows the reactivation of oxygen evolution by
reconstitution of the 23 kDa protein modified with various
concentrations of NSP. When the 33 kDa protein was
reconstituted with urea/NaCl-washed PSII membranes in
which no oxygen evolution was detected even in the
presence of CaCl
2
, the oxygen evolution was reactivated
to 0, 96 and 252 lmol O
2
Æmg chl
)1
Æh
)1
in the absence and
presence of 10 m
M
NaCl and 5 m
M
CaCl
2
, respectively. The
activity further recovered to 142 and 243 lmol O
2
Æmg
chl
)1
Æh
)1
in the absence and presence of 10 m
M
NaCl by
additional reconstitution of the unmodified 23 kDa protein,
though little effects were detected on the activity in the
presence of 5 m
M
CaCl
2
by the additional reconstitution.
When the NSP-modified 23 kDa proteins were
Fig. 2. Reconstitution of the unmodified, NSP- or GME-modified
23 kDa protein together with the 33 kDa protein with urea/NaCl-
washed PSII membranes. Urea/NaCl-washed PSII membranes were
reconstituted with the unmodified, NSP- or GME-modified 23 kDa
protein together with the 33 kDa protein. The reconstituted PSII
membranes were again treated with 2.6
M
urea, 0.2
M
NaCl and their
centrifuged supernatants were analyzed by SDS/PAGE to determine
the amounts of the 33 and 23 kDa proteins rebound after reconstitu-
tion. Lane 1, unwashed PSII-membranes; lane 2, urea/NaCl-washed
PSII membranes; lane 3, urea/NaCl–washed PSII membranes recon-
stituted with the 33 kDa protein; lane 4, urea/NaCl–washed PSII
reconstituted with the 33 kDa protein and unmodified 23 kDa protein;
lanes 5–9, urea/NaCl–washed PSII membranes reconstituted with the
33 kDa protein and the 23 kDa protein modified by 0.5 m
M
NSP
(lane 5), 1 m
M
NSP (lane 6), 2 m
M
NSP (lane 7), 4 m
M
NSP (lane 8),
and 6 m
M
NSP (lane 9); lane 10, urea/NaCl–washed PSII membranes
reconstituted with the 33 kDa protein and the GME-modified 23 kDa
protein.
Fig. 1. Isoelectric focusing of the NSP- (A) or GME- (B) modified
23 kDa protein. (A) Lane 1, unmodified 23 kDa protein; lanes 2–6, the
23 kDa protein modified by NSP at concentrations of 0.5 m
M
(lane 2),
1m
M
(lane 3), 2 m
M
(lane 4), 4 m
M
(lane 5), 6 m
M
(lane 6). (B) Lane 1,
unmodified 23 kDa protein; lane 2, the 23 kDa protein modified with
100 m
M
GME in the presence of 2 m
M
EDC at 25 °Cfor12h.
Table 1. Reactivation of oxygen evolution by reconstitution of the NSP- or GME-modified 23 kDa protein to urea/NaCl-washed PSII membranes
reconstituted with the 33 kDa protein. Values shown are the averages of three measurements. 23, 23 kDa protein; 33, 33 kDa protein.
PS II membrane treatment
Oxygen evolution [lmol O
2
Æ(mg chl)
)1
Æh
)1
]
–Ion (%) +10 mM NaCl (%) +5 mM CaCl
2
(%)
Control PSII membranes 523 ± 26 (100) 525 ± 17 (100) 535 ± 18 (100)
Urea/NaCl-washed PSII 0 ± 0 (0) 0 ± 0 (0) 0 ± 0 (0)
+ 33 0 ± 0 (0) 96 ± 7 (18) 252 ± 13 (47)
+ 33 + 23 142 ± 7 (27) 243 ± 10 (46) 274 ± 12 (51)
+ 33 + 0.5 mM NSP-modified 23 25 ± 5 (5) 120 ± 9 (23) 265 ± 11 (50)
+ 33 + 1.0 mM NSP-modified 23 13 ± 3 (2) 110 ± 7 (21) 267 ± 12 (50)
+ 33 + 2.0 mM NSP-modified 23 7 ± 2 (1) 103 ± 7 (20) 260 ± 10 (49)
+ 33 + 4.0 mM NSP-modified 23 0 ± 0 (0) 95 ± 5 (18) 263 ± 12 (49)
+ 33 + 6.0 mM NSP-modified 23 0 ± 0 (0) 94 ± 6 (18) 253 ± 10 (47)
+ 33 + GME-modified 23 140 ± 9 (27) 250 ± 9 (48) 252 ± 12 (47)
ÓFEBS 2004 Interaction between the 23 and 33 kDa proteins (Eur. J. Biochem. 271) 965
reconstituted together with the 33 kDa protein, their
reactivations in the absence and presence of 10 m
M
NaCl
decreased with increasing NSP concentrations, and no
reactivation effects were observed in PSII membranes
reconstituted with the 23 kDa protein modified with NSP
above 4 m
M
.
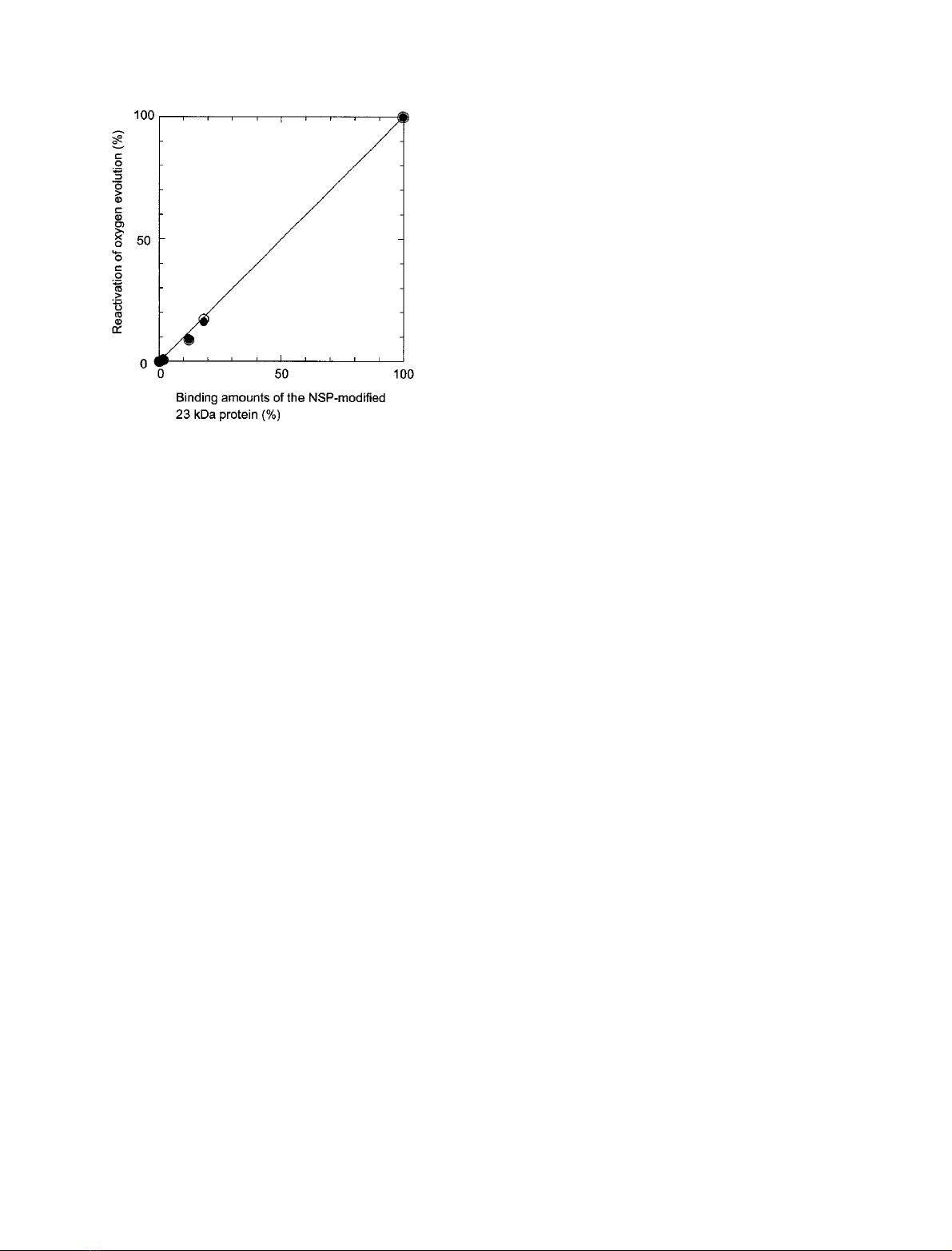
Figure 3 shows the correlation between the amounts of
rebound 23 kDa protein (Fig. 2) and reactivation of oxygen
evolution in the absence (open circles) and presence (closed
circles) of 10 m
M
NaCl (Table 1). Their good correlation
indicates that loss of the reactivating capability of the NSP-
modified 23 kDa protein was caused directly by loss of their
rebinding, which in turn suggests that the modified protein,
when rebound, are fully functional and that there is
apparently no nonspecific binding of the modified protein.
In contrast to the NSP-modified 23 kDa protein, the
GME-modified 23 kDa protein retained its capabilities to
rebind with the 33 kDa protein associated with PSII and to
reactivate the oxygen evolution. Figure 1B shows that the
pI values were upshifted from 6.8 of unmodified protein
(lane 1) to 9.2 (lane 2) by modification of carboxyl groups
with GME in the presence of EDC. This change was
estimated to result from modification of around three
negatively charged carboxyl groups to uncharged groups, as
calculated using a computer pI/Mr tool. The GME-
modified 23 kDa protein completely rebound to the
33 kDa protein associated with PSII membranes (Fig. 2,
lane 10) and its rebinding reactivated the oxygen evolution
to extents comparable with the rebinding of the unmodified
23 kDa protein (Table 1). These results clearly indicate that
surface negative charges on the 23 kDa protein do not
participate in its functional binding with the 33 kDa protein
associated with PSII membranes.
Next, we attempted to identify the lysyl residues on the
23 kDa protein modified with NSP. Both of the modified
23 kDa proteins treated with 0.5 m
M
NSP and 4 m
M
NSP
whose binding abilities were lost by about 82% and 100%,
were denatured with urea and digested with Staphylococcus
V8 protease followed by determination of the mass of
the resultant peptide fragments with mass spectroscopy.
Whether a peptide fragment can be detected by the
MALDI-TOF MS depends in some cases on the matrix
employed, three different matrices were used: They were,
a-cyano-4-hydroxycinnamic acid (CHC), 2-mercapto-
benzothiazole (MBT) and 2,5-dihydroxybenzoic acid
(DHB). This led to a more complete identification of
the peptide fragments resulting from the V8 protease
digestion of the modified 23 kDa protein. The results were
shown in Table 2 (the 23 kDa protein modified with
0.5 m
M
NSP) and Table 3 (the 23 kDa protein modified
with 4 m
M
NSP). Peptide fragments yielded could be
assigned to the known amino acid sequence within a
0.01% mass error, as shown in Tables 2 and 3. Modifi-
cation of the amino group with each NSP molecule results
in an addition of an N-propionyl group, which corres-
ponds to an increase of 56.0 Da in the molecular mass. In
the 23 kDa protein modified with 0.5 m
M
NSP, there
were 31 peptides identified ranging in mass from 703.32 to
2840.49 Da (Table 2). Of these peptides, eight lysyl
residues were identified to be modified with NSP, two
Lys between Lys11 and Lys14; one Lys among Lys27 and
Lys38; one Lys at Lys40; one Lys at Lys90 or Lys96; one
Lys at Lys143 or Lys152; two Lys between Lys166 and
Lys174 (Table 2). These modified lysyl residues were
arranged in the amino acid sequence of the 23 kDa
protein as shown in Fig. 4. This indicates that eight lysyl
residues modified with 0.5 m
M
NSP are located in
six domains, namely Lys11–Lys14, Lys27–Lys38, Lys40,
Lys90–Lys96, Lys143–Lys152, Lys166–Lys174. In the
23 kDa protein modified with 4 m
M
NSP, 32 peptides
ranging in mass from 703.33 to 2760.30 Da were identi-
fied. Of these peptides, 11 lysyl residues were identified to
be modified with NSP, which were two Lys between
Lys11 to Lys14; two Lys between Lys27 and Lys38; one
Lys at Lys40; one Lys at Lys68 or Lys69; one Lys at
Lys90 or Lys96; one Lys at Lys143 or Lys152 and three
Lys between Lys166 and Lys174 (Table 3). Ten residues
in these modified Lys were found in the six domains that
wereidentifiedtobemodifiedwith0.5m
M
NSP, as
shown in Fig. 4. Only one domain of Lys68–Lys69 was
modified uniquely with 4 m
M
NSP in addition to the six
domains.
Discussion
The present results clearly demonstrated that modification
of amino groups on the 23 kDa protein with NSP
significantly affected its rebinding ability and thus the
reactivating capability of oxygen evolution. In contrast,
modification of carboxyl groups on the protein with
GME in the presence of EDC did not affect the rebinding
and reactivation capabilities. We thus conclude that the
positive charges, but not the negative charges, on the
23 kDa protein, are important for its interaction with
PSII and in particular, the 33 kDa protein associated with
PSII.
The 23 kDa protein from spinach is composed of 186
amino acid residues including 14 Asp, 10 Glu, 20 Lys, and 3
Arg [43]. In the present study, around three carboxyl groups
Fig. 3. Relationship between the amounts of NSP-modified 23 kDa
protein rebound and oxygen evolution restored. s, oxygen evolution
in the absence of NaCl; d, oxygen evolution in the presence of
10 m
M
NaCl.
966 A. Tohri et al. (Eur. J. Biochem. 271)ÓFEBS 2004


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)