
X-ray crystal structures of Phanerochaete chrysosporium
Laminarinase 16A in complex with products from lichenin
and laminarin hydrolysis
Jonas Vasur
1
, Rie Kawai
2
, Evalena Andersson
1
, Kiyohiko Igarashi
2
, Mats Sandgren
1
,
Masahiro Samejima
2
and Jerry Sta
˚hlberg
1
1 Department of Molecular Biology, University of Agricultural Sciences, Uppsala, Sweden
2 Department of Biomaterial Sciences, Graduate School of Agriculture and Life Sciences, The University of Tokyo, Japan
Introduction
Glucan architecture is determined by the pattern of
linkages connecting the glucose units. Branching,
substituents and the degree of polymerization further
distingiuish different types of glucans [1,2]. Enzymes
such as laminarinase 16A (Lam16A) from the white-
rot fungus Phanerochaete chrysosporium exploit such
differences [3] for binding and hydrolysis of glucans
such as laminarin and lichenin (lichenan).
b-1,3-glucans are omnipresent in the natural habitat
of P. chrysosporium, which comprises fallen trees and
forest litter. b-1,3-glucans are a prominent component
of fungal cell walls and are produced to varying
degrees by plants (as callose) in response to tissue
damage. Furthermore, fungi and bacteria are often
able to produce characteristic b-glucan exopolysaccha-
rides, and P. chrysosporium itself can produce extracel-
lular b-1,3-glucan that forms a gel-like sheath at its
hyphae [4–7]. The usefulness of discriminating self
from non-self in such an environment would suggest that
P. chrysosporium has different glycoside hydrolases
Keywords
1,3(4)-b-D-glucanase; 3D protein-ligand
structure; glycoside hydrolase family 16;
laminarin; lichenin
Correspondence
J. Sta
˚hlberg, Department of Molecular
Biology, University of Agricultural Sciences,
Box 590, SE-75124 Uppsala, Sweden
Fax: +46 18 536971
Tel: +46 18 471 4590
E-mail: jerry@xray.bmc.uu.se
Note
The models and electron density maps of
enzyme–substrate complexes have been
deposited in the Protein Data Bank [51] and
in the Electron Density Server [52] with
accession codes 2W39 and 2W52
(Received 27 March 2009, revised 21 April
2009, accepted 14 May 2009)
doi:10.1111/j.1742-4658.2009.07099.x
The 1,3(4)-b-d-glucanases of glycoside hydrolase family 16 provide useful
examples of versatile yet specific protein–carbohydrate interactions. In the
present study, we report the X-ray structures of the 1,3(4)-b-d-glucanase
Phanerochaete chrysosporium Laminarinase 16A in complex with b-glucan
products from laminarin (1.6 A
˚) and lichenin (1.1 A
˚) hydrolysis. The
G6G3G3G glucan, in complex with the enzyme, showed a b-1,6 branch in
the acceptor site. The G4G3G ligand–protein complex showed that there
was no room for a b-1,6 branch in the )1or)2 subsites; furthermore, the
distorted residue in the )1 subsite and the glucose in the )2 subsite
required a b-1,3 bond between them. These are the first X-ray crystal struc-
tures of any 1,3(4)-b-d-glucanase in complex with glucan products. They
provide details of both substrate and product binding in support of earlier
enzymatic evidence.
Abbreviations
ES, enzyme–substrate; GH, glycoside hydrolase; glc, glucose; Lam16A, laminarinase 16A.
3858 FEBS Journal 276 (2009) 3858–3869 ª2009 The Authors Journal compilation ª2009 FEBS

with different specificities for different needs. As a
result of its binding specificity, Lam16A is only able to
bind to and hydrolyze certain glucans. In earlier exper-
iments [3], Lam16A was isolated when P. chrysospori-
um strain K-3 [8] was grown on laminarin. Lam16A
can hydrolyze the b-1,3-glucan curdlan, b-1,3;1,4-
mixed linkage glucans such as lichenin, and certain
b-1,6-branched-1,3-glucans such as laminarin [3]. Sub-
sequent DNA sequence analysis of the P. chrysosporium
genome [9] classifies at least 20 of the 87 glycoside
hydrolase genes [10] as members of glycoside hydrolase
family 16 (GH16) [11].
Lam16A belongs to the ‘nonspecific’ 1,3(4)-b-glu-
canase subfamily [GH16 subfamilies: XTHs, (1,3;1,4)-
b-glucanases, ‘nonspecific’ 1,3(4)-b-glucanases, (1,3)-b-
galactanases, (1-4)-b-galactanases/j-carrageenases] [12]
of the relatively diverse GH family 16, which consists
of retaining endo-glucanases and transglycosylases
with a b-jelly roll fold. ‘Nonspecific’ is nevertheless a
misleading term for Lam16A because b-1,6 branching
and b-1,4 bonds specifically define where Lam16A
hydrolyzes its substrate; ‘multivalent’ may therefore be
a better analogy. When Lam16A hydrolyzes laminarin,
it produces the b-glucans G6G3G3G (6-O-glucosyl-
laminaritriose); G3(G6)G, G3G, G3G3(G6)G and
G6G3G are not detected. Similarly, G4G3G (4-O-
glucosyl-laminaribiose) is found among lichenin
(G4G4G3)
n
hydrolysis products, but G3G4G and
G4G4G are not [3]. These hydrolysis patterns for
laminarin and lichenin are dependent on the 1,3(4)-b-
glucanase used [3,13–15].
We present two X-ray structures of Lam16A–ligand
complexes. The first structure, named P4 (1.6 A
˚reso-
lution), showed the entire G6G3G3G ligand in the
acceptor site and G3G3G (with no discernable b-1,6
branch) in the donor site. In the other Lam16A–
ligand complex, P3, the discernable G3G spanned the
)2 and )1 subsites (1.1 A
˚resolution; after co-crystal-
lization with G4G3G); both structures were solved
using the apo (free enzyme) structure [16] as a tem-
plate. This revealed a total of three differently posi-
tioned ligands in the substrate-binding cleft. We show
how the acceptor site of the substrate-binding cleft is
able to accommodate a b-1,6 branch, and how steric
obstructions at other subsites precluded such accom-
modation. Furthermore, we show the movement of a
peripheral Asn162 towards the active site and how
this extended a network of hydrogen bonds. The
essential role of water molecules in mediating hydro-
gen bonding between ligand and protein is also
elucidated.
Results
Overall structure
Two different ligand complex structures of Lam16A
were obtained: one by co-crystallization with the tri-
saccharide G4G3G, and one by soaking preformed
Lam16A crystals with the tetrasaccharide G6G3G3G
(Fig. 1). For simplicity, the complex structures are
abbreviated P3 (G4G3G) and P4 (G6G3G3G). All
occupied subsites except the )1 subsite contained
4
C
1
chair conformers. The P3 and P4 structures were
solved to 1.1 and 1.6 A
˚resolution, respectively. Statis-
tics from diffraction data processing and structure
refinement are summarized in Table 1. All 298 amino
acids were elucidated in both structures. In the P4
structure, the N-glycosylation displayed a similar
geometry to that of the apo structure. In the degly-
cosylated P3 structure, only one N-acetyl glucosamine
remained.
P3 structure
The P3 structure revealed electron density for only a
G3G disaccharide in the )2 and )1 subsites (Fig. 2C).
When contoured to levels below 0.30 e A
˚
)3
, the elec-
tron density extended beyond O4 of )2 glucose
()2glc), where the nonreducing glucose unit of
100°
+2main
+2branch
+1
+3
–4
–3
–2
–1
NC
N
C
159–162 loop
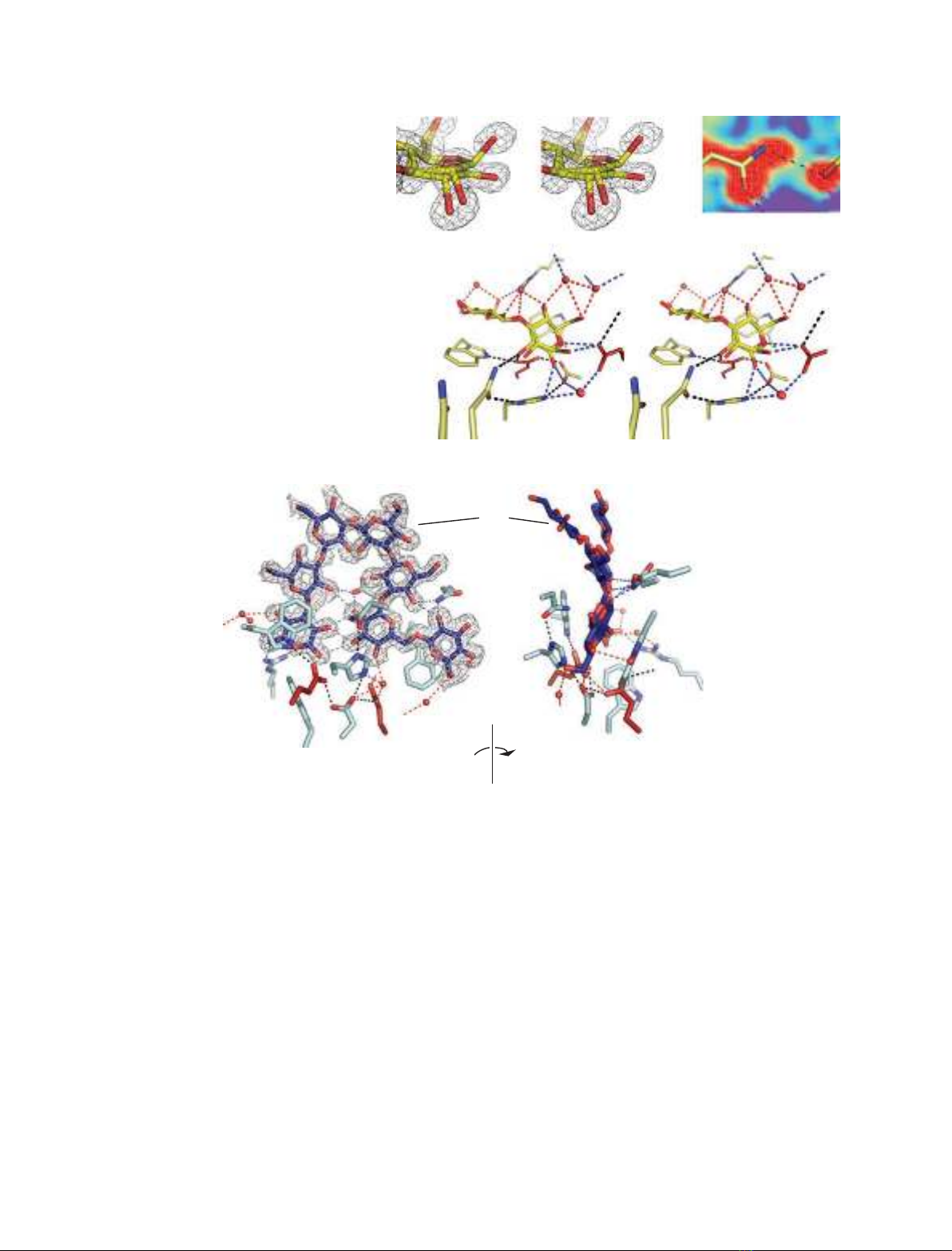
Fig. 1. Superposed Lam16A–ligand
complexes from two different angles: a
semi-transparent secondary structure and
a B-factor putty rendition. The secondary
structures are rendered in yellow for P3 and
blue for P4; the nucleophile Glu115 and
Brønsted acid/base Glu120 are shown with
white carbon atoms. The 159–162 loop is
rendered in red in the B-factor putty.
J. Vasur et al.Lam16A–product complexes
FEBS Journal 276 (2009) 3858–3869 ª2009 The Authors Journal compilation ª2009 FEBS 3859

G4G3G ligand should be, but no clear electron density
could be distinguished.
The )1 glucose residue displayed two conformers, a
1,4
Bboat conformer and a
4
Eenvelope conformer,
where O1 lay in the same plane as C5, O5, C1, C2,
C3. Gluconolactone fit the electron density better than
a distorted a-anomer (Fig. 2A). Hence, a nonrestrained
gluconolactone model was used for refinement.
The O6 and O4 hydroxyls of the ‘gluconolactone’
have especially low B-factors (5.65 and 6.75 A
˚
2
, respec-
tively; Table 1), most likely as a result of hydrogen
bonding to three conserved waters (see Experimental
procedures) deep within the substrate-binding cleft.
Conserved waters are more tightly bound than tran-
sient waters [17] and can be considered as extensions
of the peptide chain [18]. These water molecules form
a hydrogen bond network [19,20], which extends from
the Brønsted acid/base Glu120 to Arg73 Ng1
(Fig. 2C). The network continues through Arg73 Ng2
to O6 of the )2glc residue and on through two more
conserved waters, ending in the main-chain carbonyl
oxygen of Glu107.
On the other side of the G3G ligand, there is a
U-shaped chain of hydrogen bonded amino acid side
chains comprising Trp110, Glu115 (nucleophile),
Asp117, His133 and the ‘swung-in’ Asn162 ‘A’ con-
former (Fig. 2B). Angles and distances between the
nitrogens and oxygens of these side chains indicate
hydrogen bonding. The O2 of )1glc (or sugar 2-hydro-
xyl) hydrogen bonds to both Asn162 ‘swung-in’ con-
former and to Glu115 (the nucleophile). The distorted
electron density around Nd2 of Asn162 (Fig. 2B) is
probably a proton involved in this hydrogen bond.
The ring carbons of )2glc residue lie 4 A
˚away from
and parallel to the 6-ring of Trp110. Trp103, the
hydrophobic platform found at the )1 subsite in
almost all glycoside hydrolases [21], aligns the C3, C4,
C5 and C6 atoms of )1glc. Protein-ligand interactions
are listed in Table 2.
P4 structure
The unconventional binding of the b-1,6 branch deep
in the acceptor site of the P4 structure can result in
ambiguous numbering of the +2 subsites. The conven-
tional subsite numbering [22] in the acceptor site has
been modified as follows: closest to the site of catalysis
is the +1glc residue, G6G3G3G; G6G3G3G is the
‘+2glc branch’ residue, G6G3G3G is the ‘+2glc main
chain’, and G6G3G3Gis the +3glc main chain residue
(Fig. 3).
The P4 complex contains two ligand molecules
(Fig. 3). In the donor site, three b-1,3-linked glucose
residues are visible in subsites )4to)2, but no b-1,6
branch is visible beyond )4glc. The acceptor ligand
shows unambiguous electron density for all four glu-
cose units: +1, +2glc main chain, +3 and a +2glc
branch residue. The )1 subsite is unoccupied. The
exposed tryptophans Trp110 and Trp257 act as hydro-
phobic platforms that align with the glucose residues
)2glc and +2glc branch, respectively.
The substrate-binding cleft forms a straight and
narrow, open-ended canyon (Fig. 3B). Because it is
open-ended, it is easy to imagine a more linear oligo-
Table 1. Data collection, processing, refinement and model
statistics.
G4G3G G6G3G3G
(2W39) (2W52)
Data collection
Wavelength (A
˚) 0.934 1.041
Spacegroup P2
1
2
1
2
1
P2
1
2
1
2
1
Cell dimensions (A
˚) 38.1, 46.7, 152.6 38.1, 46.7, 152.9
Resolution (A
˚)
a
29.6-1.10
(1.16–1.10)
34.4-1.55
(1.63–1.55)
Unique reflections
a
98350 (7958) 39543 (4760)
Redundancy
a
3.8 (2.8) 6.5 (6.1)
Completeness (%)
a
88 (50) 97 (82)
R
meas
(%)
a,b
0.067 (0.44) 0.044 (0.13)
ÆI/r(I)æ15.4 (3.2) 35.8 (15.1)
Refinement and model statistics
Reflections in working set 93358 37489
R
final
/R
free
13.8/15.2 15.1/17.1
Number of non hydrogen
atoms
2873 2871
Amino acid 2388 2287
Ligand 35 79
Water 436 422
Glycosylation 14 83
B
Wilson
(A
˚
2
)
c
713
Mean temperature factors B
ave
(A
˚
2
)
Amino acids (1–298) 9 9
Ligand 14 24
)2glc 14 19
)1glc ‘gluconolactone’ 11 –
)1glc ‘b-anomer’ 17 –
+1glc – 16
+2branch – 21
+2main – 18
rmsd from ideal values
d
Bond distance (A
˚) 0.009 0.013
Bond angle () 1.5 1.5
Ramachandran outliers
e
T81 T81, D243,
C254, W257
a
Values for highest resolution shell in parentheses.
bRmeas ¼Phffiffiffiffiffiffiffi
nh
nh1
pPj^
IhIh;ij
PhPnh
iIh;i
where ^
Ih¼1
nhPnh
iIh;iand n
h
¼redun-
dancy [53].
c
Negative slope of B-Wilson plot obtained when scaling
in Scala [43].
d
rmsd from Engh’s and Huber’s ideal values [54].
e
As defined by AutoDep’s validation report at EBI [55].
Lam16A–product complexes J. Vasur et al.
3860 FEBS Journal 276 (2009) 3858–3869 ª2009 The Authors Journal compilation ª2009 FEBS
saccharide (such as G4G3G) lying along the length of
the unencumbered canyon floor. Nevertheless, the
ostensibly linear ligands of the G6G3G3G structure do
not lie along the floor of the canyon (Fig. 3A, bottom
left). Instead, only the b-1,6 branch extends parallel to
the floor of the cleft. The main chains, by contrast, lie
perpendicular to the canyon floor. The b-1,3 main
chains of the P4 structure are curved, resembling a
parentheses when viewed against either wall of the sub-
strate-binding cleft. A network (or chain) of bifurcated
hydrogen bonds [19,20] zig-zags between hydroxyls of
the donor and acceptor ligands, analogous to a single
shoelace lacing opposing eyelets (Fig. 3A), as if pulling
the two ligands closer toward each other. The
G6G3G3G oligosaccharide in the acceptor site also
bends out from the cleft itself, following the curvature
of the 159–162 loop, over the lip of the cleft (Fig. 3B).
Both +2glc main chain and +3glc lie 4 A
˚away from
–1
–1
–2
–2
N162
E115
E115
D117
E120
D117
E120
N162
H133 H133
R73
R73
W110
W110
W103
W103
3.3 Å
N162
–1
A
C
B
Fig. 2. The Lam16A P3 structure with a
well-defined disaccharide occupying
subsites )2 and )1. (A) Electron density
(0.35 e A
˚
)3
) at subsite )1 showing both
conformers. (B) Asn162 Nd2 (‘swung-in’
conformer) displays elongated electron den-
sity [56] towards O2 of the )1glc residue,
suggesting protonation. (C) Stereo image of
protein/ligand interactions; red dotted lines:
hydrogen bonds to conserved waters; black:
hydrogen bond network from Asn162
‘swung-in’ conformer to nucleophile Glu115;
blue: hydrogen bonds between amino acids
and ligand or between amino acids and con-
served waters.
–4
AB
–3
–2
+1
+2main
+2branch
+3
E115
–4
90°
W257
R73
W257
R73
W110
D117
E120
N162
E120
D117
H133
E115
W103
W103
Q260
D256
Fig. 3. The Lam16A P4 structure with two ligand molecules, occupying both the donor and acceptor sites of the substrate-binding cleft,
viewed (A) across and (B) along the binding cleft. Electron density (0.35 e A
˚
)3
) reveals three glucose residues ()4, )3 and )2) in the donor
site, and four residues in the acceptor site (+1, +2main, +2branch, and +3). The catalytic residues Glu115 (nucleophile) and Glu120 (acid/
base) are shown in red, other selected residues around the catalytic centre with light-blue carbon atoms, and conserved waters as red
spheres. Red dotted lines: hydrogen bonds to conserved waters; black: hydrogen bonds from and including the catalytic triad; blue: hydrogen
bonds between amino acids and ligand or between amino acids and conserved waters; gray: ligand-to-ligand hydrogen bonds.
J. Vasur et al.Lam16A–product complexes
FEBS Journal 276 (2009) 3858–3869 ª2009 The Authors Journal compilation ª2009 FEBS 3861
residues Asn162 and Gly161, which is a typical dis-
tance for van der Waals interactions. The 159–162
loop deviates markedly from that of the apo structure
and the P3 structure. Asn162 Od1 has moved by more
than 2 A
˚(relative to the ‘swung-out’ position of the
apo structure) to the ‘swung-in’ position to hydrogen
bond with His133 Nd1. Asn162 Od1 hydrogen bonds
with His133, creating the U-shaped chain of hydrogen
bonds as in the P3 structure. Also, residues of the
159–162 loop interact hydrophobically with the accep-
tor ligand. In the P4 structure, only the Asn162
‘swung-in’ conformer was detected (where Asn162
hydrogen bonds with His133).
Superposition of structures
The two enzyme–ligand complexes were superposed
onto the apo structure [16], yielding a rmsd of 0.19 A
˚
(P4) and 0.12 A
˚(P3) (values normalized to 100 amino
acid residues) [23]. Superposition of the P3, P4, and
apo structures revealed ten conserved waters within the
substrate-binding cleft. Conserved waters tended to
be in the donor site and at the base of the substrate-
binding cleft.
The P4 structure reveals how the G6G3G3G product
binds after hydrolysis. The binding differs from that
expected for a productive enzyme–substrate (ES) com-
plex with laminarin. This is seen when using the super-
posed P3 structure as a reference for binding at the )1
subsite. In a laminarin–Lam16A complex, O3 of +1glc
would be the scissile bond oxygen, covalently bound to
the anomeric carbon C1 of )1glc and within hydrogen
bond distance to the catalytic acid/base Glu120, very
close to where the anomeric oxygen O1 of the )1b-glu-
cose unit is located in the P3 structure. By contrast, the
acceptor ligand of P4 is shifted away from the canyon
floor (Fig. 4) and +1glc O3 is 2.2 A
˚away from )1glc
O1 in P3. This implies that the +1glc, and consequently
the +2glc branch, bind deeper in the cleft just before
hydrolysis of the glycosidic bond.
The position of the P4 donor ligand molecule also
deviates from that of a would-be substrate spanning
the catalytic center. When superposed with the P3
structure, the O6 atoms of )2glc overlap, but the
)2glc of P4 is pivoted by 14around O6 towards the
)1 subsite. The )2glc O1 in P4 is 1.8 A
˚away from
that in P3 (i.e. from where it would be expected in an
ES complex or the glycosyl–enzyme intermediate). The
)2glcO6 is held firmly in place by hydrogen-bonding
to Arg73 Ng2. This pivot point is the only conven-
tional (neither weak, bifurcated, nor water-mediated)
[19,20] protein–ligand hydrogen bond in the P4
structure.
Substrate models
The computed models of laminarin and lichenin (i.e.
the substrates from which the ligands are derived)
exhibit distinctly different strucutres. In b-1,3-linked
glucans such as laminarin, a six-glucose helical repeat
is evident (Fig. 5). The G4G4G3 repeats of lichenin
make a markedly straighter chain, resembling kinked
cellulose.
Comparison of the bound (G6)G3G3G ligands with
the b-1,3-glucan model (Fig. 5 and Table 3) suggests
that the curved shapes of the P4 ligands are low energy
conformations intrinsic to the ligands themselves, and
not a result of forces exerted upon them by the enzyme.
Discussion
The 1,3(4)-b-glucanases differ from other GH16
enzymes in their ability to hydrolyze distinctly differ-
ent glucans (such as lichenin and laminarin) (Fig. 5).
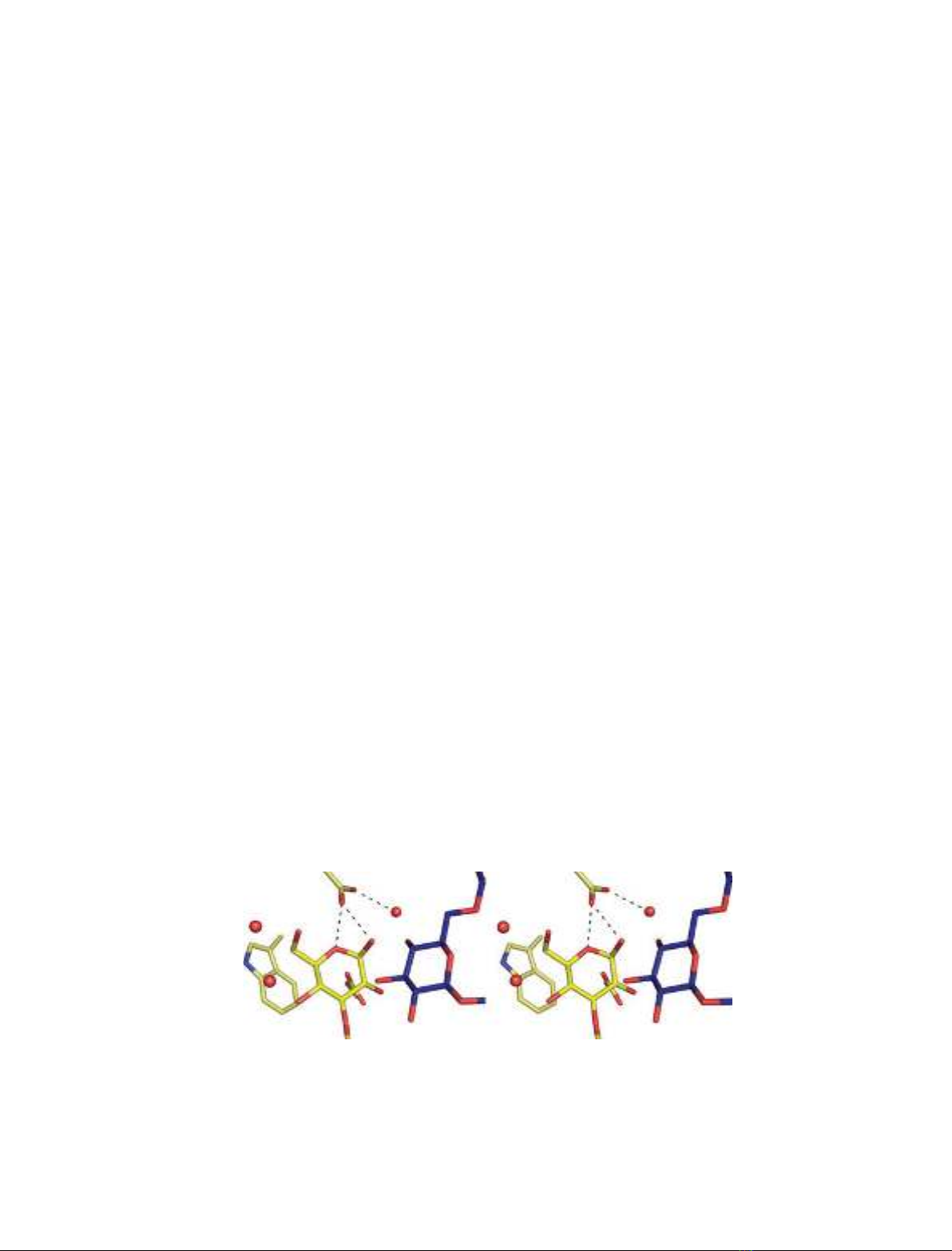
O3
E115
E120
O1
W103
--2.2 Å--
–1 +1
2.5 Å
O3
E115
E120
O1
W103
--2.2 Å--
+1
2.5 Å
–1
Fig. 4. Superposition of the P3 b-glucose conformer at subsite )1 (yellow ligand) and P4 (blue ligand) structures of Lam16A. Stereo enlarge-
ment of the ligands at the catalytic center, showing that the O3 of +1glc (blue) is in the same orientation as it would be before hydrolysis
(i.e. not flipped). Upon substrate binding, O3 would be where the anomeric oxygen of +1glc is and its O4 hydroxyl would be where the
Glu120-bound water is.
Lam16A–product complexes J. Vasur et al.
3862 FEBS Journal 276 (2009) 3858–3869 ª2009 The Authors Journal compilation ª2009 FEBS


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)