
Kinetic studies on endo-b-galactosidase by a novel colorimetric assay
and synthesis of
N
-acetyllactosamine-repeating oligosaccharide
b-glycosides using its transglycosylation activity
Takeomi Murata, Takeshi Hattori, Satoshi Amarume, Akiko Koichi and Taichi Usui
Department of Applied Biological Chemistry, Shizuoka University, Japan
Novel chromogenic substrates for endo-b-galactosidase
were designed on the basis of the structural features of
keratan sulfate. Galb1-4GlcNAcb1-3Galb1-4GlcNAcb-
pNP (2), which consists of two repeating units of N-acetyl-
lactosamine, was synthesized enzymatically by consecutive
additions of GlcNAc and Gal residues to p-nitrophenyl
b-N-acetyllactosaminide. In a similar manner, Glc-
NAcb1-3Galb1-4GlcNAcb-pNP (1), GlcNAcb1-3Galb1-
4Glcb-pNP (3), Galb1-4GlcNAcb1-3Galb1-4Glcb-pNP (4),
Galb1-3GlcNAcb1-3Galb1-4Glcb-pNP (5), and Galb1-
6GlcNAcb1-3Galb1-4Glcb-pNP (6) were synthesized as
analogues of 2. Endo-b-galactosidases released GlcNAcb-
pNP or Glcb-pNP in an endo-manner from each substrate.
A colorimetric assay for endo-b-galactosidase was devel-
oped using the synthetic substrates on the basis of the
determination of p-nitrophenol liberated from GlcNAcb-
pNP or Glcb-pNP formed by the enzyme through a coupled
reaction involving b-N-acetylhexosaminidase (b-NAHase)
or b-
D
-glucosidase. Kinetic analysis by this method showed
that the value of V
max
/K
m
of 2for Escherichia freundii endo-
b-galactosidase was 1.7-times higher than that for keratan
sulfate, indicating that 2is very suitable as a sensitive sub-
strate for analytical use in an endo-b-galactosidase assay.
Compound 1still acts as a fairly good substrate despite the
absence of a Gal group in the terminal position. In addition,
the hydrolytic action of the enzyme toward 2was shown to
be remarkably promoted compared to that of 4by the
presence of a 2-acetamide group adjacent to the p-nitro-
phenyl group. This was the same in the case of a comparison
of 1and 3. Furthermore, the enzyme also catalysed a
transglycosylation on 1and converted it into GlcNAc
b1-3Galb1-4GlcNAcb1-3Galb1-4GlcNAcb-pNP (9)and
GlcNAcb1-3Galb1-4GlcNAcb1-3Galb1-4GlcNAcb1-3Galb1-
4GlcNAcb-pNP (10) as the major products, which have
N-acetyllactosamine repeating units.
Keywords: endo-b-galactosidase; enzyme assay; kinetics;
poly (N-acetyllactosamine); transglycosylation.
Endo-b-galactosidases were discovered as keratan sulfate-
degrading enzymes, so-called keratanases, in culture filtrates
of Escherichia freundii (glycoside hydrolase family 16) [1],
Coccobacillus sp. [2], Pseudomonas sp. [3], Flavobacterium
keratolyticus (glycoside hydrolase family 16) [4,5], and
Bacteroides fragilis [6]. E. freundii keratanase was found to
have hydrolysing activity for a wide range of nonsulfated
oligosaccharides isolated from human milk and carbohy-
drate moieties of glycoproteins and glycolipids [7–10]. The
use of endo-b-galactosidase has been expanded to detection
of poly (N-acetyllactosamine) chains in a variety of complex
glycoconjugates in addition to keratan sulfate. Bacteroides
fragilis endo-b-galactosidase has properties similar to those
of E. freundii endo-b-galactosidase [11–13]. Therefore, the
endo-b-galactosidases from E. freundii and B. fragilis have
been widely used as tools for structural and functional
analyses of glycans involved in glycoconjugates.
An assay using keratan sulfate as a substrate has been
widely used for estimation of endo-b-galactosidase activity.
However, this method is not always reproducible because
of lack of uniformity of the polymer. Methods using
low molecular mass substrates have been preferred and
recommended for accurate determination of activities of
endoglycosidases such as a-amylase [14], lysozyme [15],
and endo-b-N-acetylglucosaminidase [16], because the
purity of the substrate and the reaction pattern can be
determined exactly. This led us to develop a substrate
suitable for use in the analysis of endo-b-galactosidase. A
series of chromogenic substances having a partially substi-
tuted unit of poly (N-acetyllactosamine) were designed as
substrate analogues for this enzyme because systematic
kinetic studies on the structural modification of substrates
would be helpful in revealing the requirements for binding
and catalytic specificity. In general, glycosidase can cause
transglycosylation as well as hydrolysis as a reverse
reaction [17–19]. Transglycosylation of endo-type glycosi-
dase is now used for the synthesis of N-acetyllactosamine-
repeating oligosaccharide.
Correspondence to T. Murata, Department of Applied Biological
Chemistry, Shizuoka University, 836 Ohya, Shizuoka, 422-8529,
Japan. Fax and Tel.: +81 54 238 4872,
E-mail: actmura@agr.shizuoka.ac.jp
Abbreviations:pNP, p-nitrophenyl; b-NAHase, b-N-acetylhexos-
aminidase; b4GalT, b-1,4-galactosyltransferase; b3GnT, b-1,3-N-
acetylglucosaminyltransferase; HPAEC-PAD, high-performance
anion exchange chromatography-pulsed amperometric detection.
Enzymes: endo-b-galactosidase (EC 3.2.1.103); b-
D
-galactosidase
(EC 3.2.1.23); b-1,4-galactosyltransferase (EC 2.4.1.22); b-1,3-N-
acetylglucosaminyltransferase (EC 2.4.1.149); b-D-glucosidase
(EC 3.2.1.21); b-N-acetylhexosaminidase (EC 3.2.1.52).
(Received 20 December 2002, revised 11 July 2003,
accepted 17 July 2003)
Eur. J. Biochem. 270, 3709–3719 (2003) FEBS 2003 doi:10.1046/j.1432-1033.2003.03757.x

In this paper, we describe the enzymatic synthesis of a
novel substrate 2and its analogues for use a colorimetric
assay of endo-b-galactosidase activity and the usefulness of
the resulting chromogenic substrates for kinetic studies on
the enzyme. In the latter part of this paper, synthesis of
N-acetyllactosamine-repeating oligosaccharide b-glycoside
utilizing endo-b-galactosidase-mediated transglycosylation
is described.
Materials and methods
Materials
Endo-b-galactosidases from E. freundii and B. fragilis were
from Seikagaku Corporation (Tokyo, Japan) and Wako
Pure Chemical Industries, Ltd. (Osaka, Japan), respectively.
b-
D
-Galactosidase from Bacillus circulans ATCC31382 [20]
was a kind gift from Meiji Milk Products Co. Ltd. (Tokyo,
Japan). b-
D
-Glucosidase from almonds was from Sigma
Chemicals. b-N-acetylhexosaminidase (b-NAHase) from
Amycolatopsis orientalis IFO 12806T was purified by 80%
saturated ammonium sulfate precipitation followed by
GlcNAc-cellulofine affinity chromatography. Bovine milk
b-1,4-galactosyltransferase (b4GalT) was from Calbiochem
(CA, USA). Crude b-1,3-N-acetylglucosaminyltransferase
(b3GnT) from bovine serum was prepared as follows.
Bovine serum was brought to 80% saturation with solid
ammonium sulfate and left standing overnight at 4 C. The
precipitate was collected by centrifugation at 5000 gfor
30 min, dissolved in 50 m
M
Tris/HCl buffer (pH 8.0), and
dialysed against distilled water overnight at 4 C. The
enzyme solution was lyophilized and then used for synthesis
of oligosaccharides without further purification. The crude
enzyme preparation catalysed the transfer of a GlcNAc
residue from UDP-GlcNAc to the OH-3¢positions of
Galb1-4Glc, Galb1-4GlcNAc, Galb1-4Glcb-pNP and
Galb1-4GlcNAcb-pNP. The specific activity for Galb1-
4GlcNAcb-pNP as an acceptor substrate was 69 lUÆmg
)1
.
Galb1-4GlcNAc, GlcNAcb1-3Galb1-4GlcNAc, Galb1-
4Glcb-pNP, Galb1-4GlcNAcb-pNP, and GlcNAcb1-
6Galb1-4Glcb-pNP were synthesized by our previously
described methods [21–24]. UDP-GlcNAc and UDP-Gal
were kind gifts from Yamasa Corporation (Choshi,
Japan). All other chemicals were obtained from commercial
sources.
Enzyme assay
b-
D
-Galactosidase, b-
D
-glucosidase and b-NAHase activit-
ies were assayed as follows. A mixture containing 2 m
M
substrate solution (Galb-pNP, Glcb-pNP, and GlcNAcb-
pNP) in 0.4 mL 50 m
M
sodium phosphate buffer pH 6.0
and an appropriate amount of enzyme in a total volume of
0.1 mL was incubated for 10 min at 40 C. One hundred
microlitres of the reaction mixture were then added to
0.1 mL 1.0
M
Na
2
CO
3
on a microplate at 2-min intervals
during to stop the reaction, and the amount of liberated
p-nitrophenol was determined by measuring absorbance at
405 nm using a microplate reader (Biolumin 960, Amer-
sham Pharmacia). One unit of enzyme was defined as the
amount releasing 1 lmol p-nitrophenolÆmin
)1
.Theb3GnT
assay was carried out as follows. Galb1-4GlcNAcb-pNP
(5 mg) and UDP-GlcNAc (3.6 mg) were dissolved in
0.5mL50m
M
Tris/HCl buffer pH 8.0 containing 1.6 mg
MnCl
2
and 0.5 mg ATP, followed by addition of an
appropriate amount of b3GnT preparation. The reaction
mixture was incubated at 37 C for 96 h. Fifty microlitres of
the reaction mixture was taken out at 24-h intervals during
the reaction and boiled for 5 min. Resulting GlcNAcb1-
3Galb1-4GlcNAcb-pNP was measured by HPLC as des-
cribed in the Analytical methods.
Analytical methods
HPLC was carried out using a Mightysil RP18ODS column
(4.6 ·150 mm, Kanto Chemical Co. Ltd, Tokyo, Japan) in
a Hitachi 6000-series liquid chromatograph with an L-4000
ultraviolet detector (absorbance at 300 nm). Elution of the
column was performed with H
2
O/CH
3
OH (95 : 5, v/v). The
flow rate was 1.0 mLÆmin
)1
at 40 C. HPAEC-PAD
analysis was conducted on a DX-300 Bio-LC system
equipped with a pulsed amperometric detector (Dionex,
Sunnyvale, USA). Oligosaccharides were analysed by a
CarboPac P-1 column (Dionex, 4 ·250 mm) at a flow rate
of 1 mLÆmin
)1
at room temperature. The elution was
effected with 100 m
M
NaOH for 40 min. For NMR
analysis, an appropriate oligosaccharide sample was dis-
solved in 200 lLofD
2
O and filtrated with a Millipore filter
(0.22 lm) and then put into a sample tube (i.d. 3 mm).
1
H- and
13
C-NMR spectra were recorded on a JEOL
JNM-LA 500 spectrometer at 25 C. Chemical shifts are
expressed in drelative to sodium 3-(trimethylsilyl) propio-
nate as an external standard. ESI-MS analysis was carried
out in the positive-ion mode on a JEOL MS-700 (JEOL
Ltd, Akishima, Japan) using 2,5-dihydroxy benzoic acid as
the matrix. Determination of the amount of protein was
carried out using a Bio-Rad protein assay kit. Determin-
ation of total carbohydrate was carried out as follows. One
hundred microliters of sample was put into a test tube
(1 ·10 cm), and 100 lL of 5% (w/v) phenol and 0.5 mL
concentrated sulfuric acid were immediately added. The
sample mixture was vortexed and then kept for 20 min at
room temperature, and absorbance was read at 490 nm.
Preparation of GlcNAcb1-3Galb1-4GlcNAcb-
p
NP (1)
and GlcNAcb1-3Galb1-4GlcNAcb-
p
NP (3)
Galb1-4GlcNAcb-pNP (504 mg, 1 mmol) and UDP-Glc-
NAc (651 mg, 1 mmol) were dissolved in 25 mL 50 m
M
Tris/HCl buffer pH 8.0 containing 99 mg MnCl
2
, followed
by addition of 850 mU of crude b3GnT preparation
from bovine serum. The mixture was incubated for 172 h
at 37 C, and the reaction was terminated by boiling for
5 min. The precipitate was removed by centrifugation
(8 000 g, 20 min), and the supernatant was loaded onto a
Toyopearl HW-40S column (5 ·100 cm) equilibrated with
25% methanol at a flow rate of 2 mLÆmin
)1
.Afterthe
chromatography, the eluate was monitored by measuring
the absorbance at 300 nm (p-nitrophenyl group) and at
490 nm (phenol-sulfuric acid method) by a spectrometer.
Both chromatograms showed two peaks (F-1, tubes 24–33;
F-2, tubes 49–60). F-1 was combined, concentrated, and
lyophilized to produce 1(24.1 mg) in a 3.4% total
yield based on the acceptor added. F-2 was recovered as
3710 T. Murata et al. (Eur. J. Biochem. 270)FEBS 2003

Galb1-4GlcNAcb-pNP (0.35 g). In the same way, com-
pound 3was prepared from Galb1-4Glcb-pNP (0.5 g) and
UDP-GlcNAc (450 mg) by use of bovine serum b3GnT in
a 4% total yield based on the acceptor added.
1
Hand
13
C-NMR data of 1and 3were almost identical to data
reported previously [24].
Preparation of GlcNAcb1-3Galb1-4GlcNAcb-
p
NP (2)
and GlcNAcb1-3Galb1-4GlcNAcb-
p
NP (4)
Compound 1(24.1 mg, 34.1 lmol) and UDP-Gal (42.6 mg,
68.2 lmol) were dissolved in 3.4 mL 100 m
M
sodium
cacodylate buffer (pH 6.8) containing 67.3 mg MnCl
2
followed by addition of 0.2 U b4GalT from bovine milk.
The mixture was incubated for 6 h at 37 C and separated
by a Toyopearl HW-40S column (2.5 ·80 cm) as described
above. Compound 2was obtained in an 82% total yield
(24.3 mg) based on the acceptor added. In the same
way, compound 4was obtained in a 71% total yield
(13.2 mg) based on the acceptor from 3and UDP-Gal.
1
Hand
13
C-NMR data of 2and 4are summarized in
Table 1.
Preparation of GlcNAcb1-3Galb1-4GlcNAcb-
p
NP (5) and
its positional isomer GlcNAcb1-3Galb1-4GlcNAcb-
p
NP (6)
Compound 3(85 mg, 128 lmol) and o-nitrophenyl b-
D
-
galactopyranoside (Galb-oNP, 238 mg, 790 lmol) were
dissolved in 5.8 mL 40 m
M
sodium acetate buffer pH 5.5
followed by addition of 98 mU of b-
D
-galactosidase from
Bacillus circulans ATCC31382. The mixture was incubated
for 10 h at 50 C and was loaded onto an ODS DM1020T
column (5 ·100 cm) equilibrated with 5% methanol at
a flow rate of 5 mLÆmin
)1
in order to remove the
o-nitrophenol liberated during the reaction. The fractions
showing absorbance at 300 nm were concentrated and
loaded onto a Toyopearl HW-40S column as above. The
chromatogram showed two peaks (F-1, 1340–1520 mL;
F-2, 1640–1880 mL). F-2 contained 3(43 mg) used as the
acceptor substrate. F-1 was further separated by a Shodex
Asahipak NH2P-50 column (21.5 ·300 mm) equilibrated
with 80% acetonitrile at a flow rate of 5 mLÆmin
)1
at 40 C.
Eluate was monitored on-line by measuring the absorbance
at 300 nm. The chromatogram showed two peaks (F-1a,
327–390 mL; F-1b, 396–450 mL). These peaks were
Table 1.
1
H- and
13
C-chemical shifts of transfer products in D
2
O solution. J
1,2
, coupling constants are given in Hz.
Compounds
Chemical Shifts (d)
C-1 C-2 C-3 C-4 C-5 C-6 NHCOCOCH
3
o-ph m-ph p-ph C-O H-1 J
1,2
CH
3
2pNP 119.3 128.9 145.5 164.4
GlcNAc 101.3 57.6 74.8 80.9 77.9 62.6 177.7 24.8 5.35 8.5 2.02
Gal 105.7 72.7 84.9 71.1 77.7 63.81 4.51 8.3
GlcNAc 105.63 58.0 75.0 80.8 77.3 62.5 177.7 24.9 4.72 8.3 2.05
Gal 105.58 73.7 75.3 71.3 78.1 63.77 4.49 8.2
4pNP 119.3 128.9 145.5 164.5
Glc 102.0 75.2 77.9 80.7 76.9 62.7 5.31 7.7
Gal 105.8 72.8 84.9 71.2 77.7 63.7 4.486 7.7
GlcNAc 105.7 58.0 77.9 81.0 77.4 62.6 177.7 25.0 4.72 8.2 2.05
Gal 105.6 73.8 76.9 71.4 78.2 63.8 4.490 7.7
5pNP 119.2 128.9 145.4 164.4
Glc 102.0 75.2 77.9 80.6 76.8 62.6 5.30 8.0
Gal 105.4 72.8 84.79 71.1 77.7 63.77 4.47 7.9
GlcNAc 105.7 57.5 84.83 71.2 78.0 63.3 177.8 25.0 4.73 8.5 2.02
Gal 106.3 73.5 75.3 71.3 78.1 63.82 4.44 7.7
6pNP 119.3 128.9 145.4 164.4
Glc 102.0 75.2 77.9 80.6 76.8 62.6 5.30 8.0
Gal 105.74 72.8 84.8 71.1 77.7 63.78 4.47 7.7
GlcNAc 105.71 58.94 76.3 72.3 77.4 71.3 177.8 25.0 4.69 8.6 2.02
Gal 106.36 73.5 75.5 71.4 78.0 63.81 4.44 8.0
7pNP 118.2 127.3 143.3 164.0
Gal 101.8 72.1 83.4 68.6 77.1 61.7 5.20 6.5
GlcNAc 103.8 57.9 75.4 70.6 78.5 62.6 171.8 24.7 4.78 8.1 2.06
8pNP 118.2 127.5 143.2 164.2
Gal 102.3 72.2 75.1 70.4 78.4 70.1 5.22 7.6
GlcNAc 103.2 57.1 74.5 71.5 78.7 62.8 170.7 24.8 4.55 8.4 1.78
9pNP 119.3 128.9 145.5 164.5
GlcNAc 101.3 57.6 74.8 81.0 78.0 62.7 177.7 24.9 5.34 8.3 2.01
Gal 105.8 72.8 84.9 71.2 77.7 63.8 4.49 8.0
GlcNAc 105.67 58.0 75.0 80.9 77.4 62.6 177.7 25.0 4.70 8.2 2.03
Gal 105.58 72.8 84.8 71.2 77.7 63.8 4.46 8.0
GlcNAc 105.71 58.5 76.4 72.5 78.5 63.3 177.8 25.0 4.67 8.6 2.03
FEBS 2003 Kinetic studies on endo-b-galactosidase (Eur. J. Biochem. 270) 3711

combined, concentrated, and lyophilized to produce 5
(5.5 mg) and 6(7.2 mg) in 5.2 and 6.8% yields based on the
acceptor added, respectively.
1
H- and
13
C-NMR data of 5
and 6are summarized in Table 1.
Preparation of GlcNAcb1-3Galb1-4GlcNAcb-
p
NP (7)
and GlcNAcb1-3Galb1-4GlcNAcb-
p
NP (8)
Galb-pNP (390 mg, 1.29 mmol) and N, N¢-diacetylchito-
biose (GlcANc
2
, 531 mg, 1.25 mmol) were dissolved in
7.5mL20m
M
sodium acetate buffer pH 5.0 followed by
addition of 8.7 U of A. orientalis b-N-acetylhexosamini-
dase. The mixture was incubated for 100 h at 40 C, and the
reaction was terminated by boiling for 5 min. The precipi-
tate was removed by centrifugation (8 000 g, 15 min), and
the supernatant was loaded onto a Toyopearl HW-40S
column (5 ·100 cm) as above. Eluate was monitored by
measuring the absorbance at 300 nm (p-nitrophenyl group)
and at 490 nm (phenol-sulfuric acid method). The chroma-
togram showed four peaks (F-1, 135–150 mL; F-2, 190–
225 mL; F-3, 275–295 mL; F-4, 420–470 mL). F-2 and F-3
were combined, concentrated, and lyophilized to produce 8
(33.8 mg) and 7(8.6 mg), respectively, in a 6.5% total yield
based on the donor added.
1
H- and
13
C-NMR data of these
disaccharides are summarized in Table 1.
Hydrolytic actions of endo-b-galactosidase
on
p
-nitrophenyl b-glycosides
The hydrolytic actions of endo-b-galactosidase on p-nitro-
phenyl oligosaccharide b-glycosides and a reducing oligo-
saccharide listed in Table 2 were investigated by incubating
a mixture (50 lL) containing 1 m
M
of substrates in 50 m
M
sodium acetate buffer pH 5.8 with 1 mU of the enzymes at
37 C for 20 min. The enzyme hydrolysates were analysed
by HPLC or HPAEC-PAD as described in the Analytical
method section. The reaction was linear from 5 to 15 min.
Therateofattackon2was arbitrarily set at 100.
Colorimetric assay of endo-b-galactosidase activity
A mixture containing 0.5 m
M
of each p-nitrophenyl oligo-
saccharide b-glycoside and 50 mU of b-
D
-glucosidase or
25 mU of b-N-acetyllactosaminide in 500 mL 50 m
M
sodium acetate buffer pH 5.8 and an appropriate amount
of the enzyme was incubated at 37 C. Samples (each
50 lL) were taken at intervals (0, 5, 10, 15 and 20 min)
during the incubation and inactivated by adding 50 lL
1.0
M
Na
2
CO
3
. The amount of liberated p-nitrophenol was
determined by measuring absorbance at 405 nm using a
microplate reader. One unit of the enzyme was defined as
the amount hydrolysing 1 lmol of 2per min. The initial
rates of the enzymatic reaction were evaluated from kinetic
curves of product accumulation as described above. The
parameters of Michaelis–Menten-type kinetics were evalu-
ated by 1/v–1/[S] plots and the least-squares method. The
substrate concentration ranges used for compounds 1, 2, 3,
4 and 5 were 0.05–0.4, 0.02–0.75, 0.1–0.8, 0.25–2.0 and 0.25–
1.5 m
M
, respectively.
Assay of endo-b-galactosidase activity by HPLC
The standard assay was carried out as follows. A reaction
mixture (500 lL) containing an appropriate substrate and
endo-b-galactosidase in 10 m
M
sodium acetate buffer
(pH 5.8) was incubated at 37 C, and samples (each
50 lL) were taken at 3-min intervals during incubation.
After inactivation of each sample by adding 150 mL of 1
M
acetic acid, the amount of liberated GlcNAcb-pNP was
determined by HPLC as described in Analytical methods.
Transglycosylation reaction of endo-b-galactosidase
from
E. freundii
Compound 1(16 mg, 23 lmol) was dissolved in 1.9 mL
20 m
M
sodium acetate buffer pH 5.8 followed by addition
of 2.3 mU endo-b-galactosidase from E. freundii.The
mixture was incubated for 30 days at 37 C and was loaded
onto a Sep-pak accel QMA column (2 ·4 cm) equilibrated
with H
2
O at a flow rate of 1 mLÆmin
)1
. Eluate was collected
in 2-mL fractions and monitored by measuring the absorb-
ance at 300 nm using a spectrometer. The fractions showing
absorbance at 300 nm were combined, concentrated and
loaded onto a Shodex Asahipak GS-220FP column
(7.6 ·250 mm) equilibrated with H
2
O at a flow rate of
0.6 mLÆmin
)1
at 40 C. Eluate was monitored on-line by
measuring the absorbance at 300 nm. The chromatogram
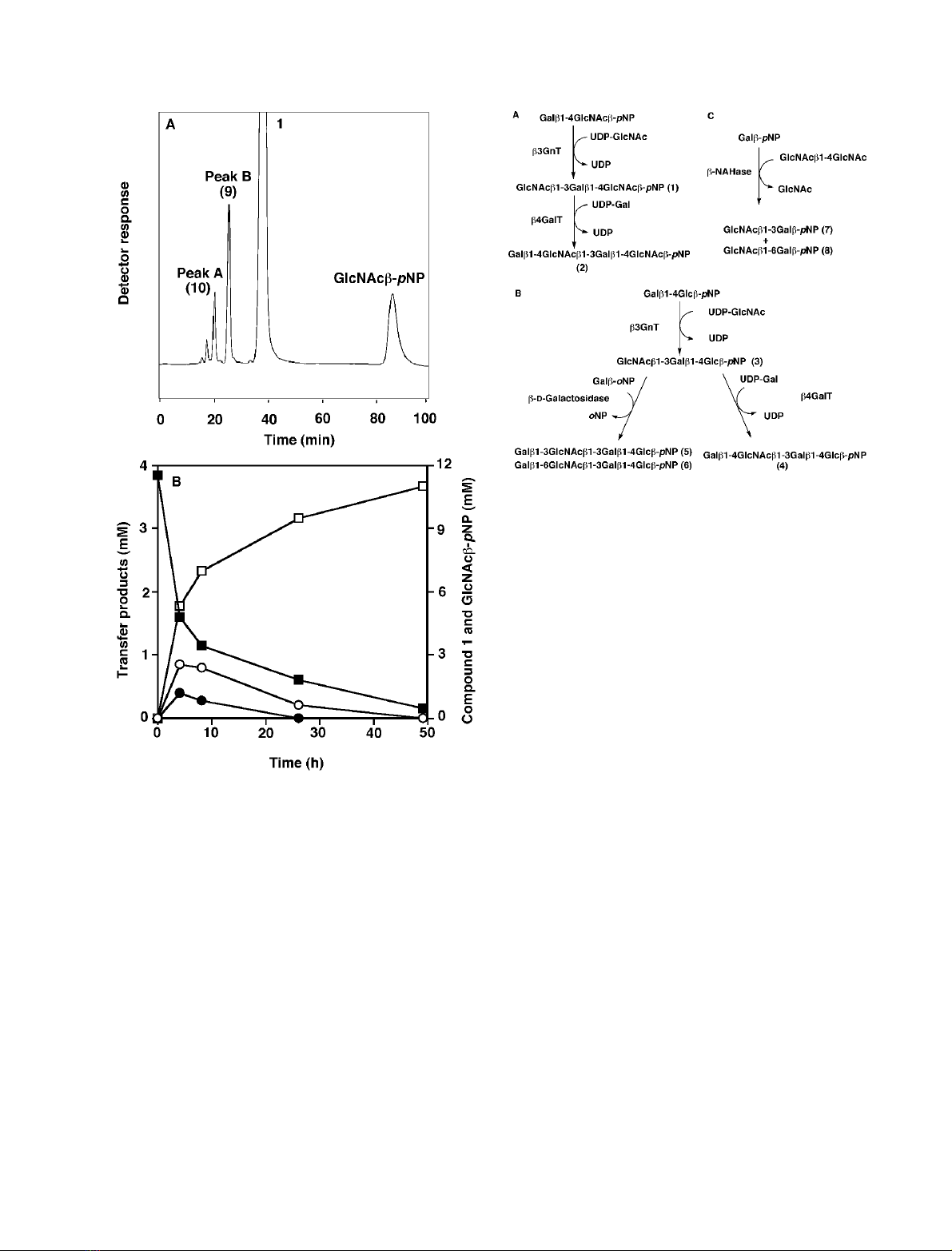
showed four peaks (Fig. 1A). Peak B, which was presumed
to be a transglycosylation product, was concentrated and
lyophilized to produce 9(0.8 mg) in 3.3% yield based on the
substrate added.
1
H- and
13
C-NMR data of 9are summar-
ized in Table 1.
Results
Preparation of colorimetric substances
A series of chromogenic substances were designed as
substrates of endo-b-galactosidase based on the structural
features of keratan sulfate, which is an alternating polymer
of N-acetyllactosamine units jointed to each other by a
Table 2. Relative hydrolytic rates of endo-b-galactosidases on p-nitro-
phenyl oligosaccharide b-glycosides. The hydrolytic actions of endo-
b-galactosidase on different substrates were investigated as described in
Materials and methods. The vertical arrow indicates the point of
cleavage. One mM of each substrate was used for the determination of
relative hydrolytic rates. –, not hydrolyzed even in the presence of 10
mU of the enzyme.
3712 T. Murata et al. (Eur. J. Biochem. 270)FEBS 2003
b-(1-3) linkage. Tetrasaccharide 2containing two N-acetyll-
actosamine repeats and its analogues were synthesized by
the alternative addition of b-(1-3) linked GlcNAc and b-(1-
4) linked Gal to Galb1-4GlcNAcb-pNP and Galb1-4Glcb-
pNP, respectively, using two kinds of glycosyltransferases.
Thus, compounds 1and 3were first prepared by the
regioselective transfer of GlcNAc residue from UDP-
GlcNAc to Galb1-4GlcNAcb-pNP and Galb1-4Glcb-pNP
by b3GnT from bovine serum. They were further converted
into 2and 4utilizing b4GalT from bovine milk (Fig. 2A,B).
The enzyme efficiently catalysed the transfer of a Gal moiety
to the OH-4¢position of the acceptors in high yields (82 and
71%) depending on the acceptor. The positional isomers 5
and 6were prepared simultaneously by Gal transfer from
Galb-oNP to the OH-3¢and OH-6¢positions of 3using
B. circulans b-
D
-galactosidase-mediated transglycosylation
(Fig. 2B). The resulting products were obtained in a molar
ratioof1:1.3andina12%overallyieldbasedonthe
acceptor added. Compound 7and its isomer 8were
prepared from Galb-pNP using b-NAHase-mediated trans-
glycosylation (Fig. 2C).
Hydrolytic actions of endo-b-galactosidases
The hydrolytic actions of endo-b-galactosidases on synthetic
chromogenic substances (each 1 m
M
) were investigated by
using enzyme preparations from E. freundii and B. fragilis.
Each enzyme splits compounds 1–6 into the corresponding
reducing oligosaccharides and chromogenic substances,
GlcNAcb-pNP/Glcb-pNP. For example, compound 2was
completely hydrolysed in an endo-manner into Galb1-
4GlcNAcb1-3Galband GlcNAcb-pNP. The relative hydro-
lytic rates of 1and 4compared with that of 2(set at 100)
were 47 and 10, i.e. 2- and 10-fold differences, respectively.
The hydrolytic activities toward 3,5and 6were not detected
under the experimental conditions as described in the
Materials and methods. Furthermore, the hydrolytic rate of
reducing trisaccharide GlcNAcb1-3Galb1-4GlcNAc was
compared with that of its glycoside 1in order to examine
how the p-nitrophenyl group participates in the hydrolytic
action. There was little progression of hydrolysis of the
reducing trisaccharide under the experimental conditions,
Fig. 1. HPLC analysis of the reaction mixture obtained by endo-b-
galactosidase-mediated transglycosylation and time courses of the pro-
duction of transglycosylation products 9 and 10 from 1 and degradation
of 1. (A) HPLC analysis was performed as described in Materials and
methods. (B) A reaction mixture (50 lL) containing 11.5 m
M
com-
pound 1, 0.1% BSA and 1.5 mU E. freundii endo-b-galactosidase in
20 m
M
sodium acetate buffer, pH 5.8, was incubated at 37 C. The
amount of each product formed from the initial substrate was deter-
mined by HPLC. s, Peak B (compound 9); d, peak A (compound 10);
h, GlcNAcb-pNP; j, compound 1.
Fig. 2. Summary of enzymatic synthesis of p-nitrophenyl oligosaccha-
ride b-glycosides used in this work. (A) Consecutive additions of Glc-
NAc and Gal to Galb1-4GlcNAcb-pNP by b3GnT and b4GalT. (B)
Consecutive additions of GlcNAc and Gal to Galb1-4Glcb-pNP by
b3GnT and b-
D
-galactosidase or b4GalT. (C) N-acetylglucosaminy-
lation of Galb-pNP by b-N-acetylhexosaminidase-mediated transgly-
cosylation.
FEBS 2003 Kinetic studies on endo-b-galactosidase (Eur. J. Biochem. 270) 3713


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)