
Substrate recognition by three family 13 yeast a-glucosidases
Evaluation of deoxygenated and conformationally biased isomaltosides
Torben P. Frandsen
1,
*, Monica M. Palcic
2
and Birte Svensson
1
1
Department of Chemistry, Carlsberg Laboratory, Copenhagen Valby, Denmark;
2
Department of Chemistry,
University of Alberta, Edmonton, Canada
Important hydrogen bonding interactions between substrate
OH-groups in yeast a-glucosidases and oligo-1,6-glucosidase
from glycoside hydrolase family 13 have been identi®ed by
measuring the rates of hydrolysis of methyl a-isomaltoside
and its seven monodeoxygenated analogs. The transition-
state stabilization energy, DDGà, contributed by the indi-
vidual OH-groups was calculated from the activities for
the parent and the deoxy analogs, respectively, according
to DDGà±RT ln[(V
max
/K
m
)
analog
/(V
max
/K
m
)
parent
]. This
analysis of the energetics gave DDGàvalues for all three
enzymes ranging from 16.1 to 24.0 kJámol
)1
for OH-2¢,-3¢,
-4¢,and-6¢, i.e. the OH-groups of the nonreducing sugar ring.
These OH-groups interact with enzyme via charged hydro-
gen bonds. In contrast, OH-2 and -3 of the reducing sugar
contribute to transition-state stabilization, by 5.8 and
4.1 kJámol
)1
, respectively, suggesting that these groups
participate in neutral hydrogen bonds. The OH-4 group is
found to be unimportant in this respect and very little or no
contribution is indicated for all OH-groups of the reducing-
end ring of the two a-glucosidases, probably re¯ecting their
exposure to bulk solvent. The stereochemical course of
hydrolysis by these three members of the retaining family 13
was con®rmed by directly monitoring isomaltose hydrolysis
using
1
H NMR spectroscopy. Kinetic analysis of the
hydrolysis of methyl 6-S-ethyl-a-isomaltoside and its 6-R-
diastereoisomer indicates that a-glucosidase has 200-fold
higher speci®city for the S-isomer. Substrate molecular rec-
ognition by these a-glucosidases are compared to earlier
®ndings for the inverting, exo-acting glucoamylase from
Aspergillus niger and a retaining a-glucosidase of glycoside
hydrolase family 31, respectively.
Keywords: protein-carbohydrate interaction; NMR; glyco-
sidase mechanism; substrate analogs; molecular recognition.
Strong intermolecular hydrogen bonds are very important
in speci®city of enzymes and other proteins that metabolize
or bind carbohydrates [1±6]. Substrate analogs such as
deoxygenated sugars, facilitate identi®cation of critical
contacts and enable quanti®cation of the energetics of the
protein±carbohydrate binding at the level of individual
interacting sugar OH-groups and functional atoms or
groups in the protein [4,7±11]. Alternatively, site-speci®c
mutants of a protein are useful in evaluation of speci®c
protein±carbohydrate interactions and further insight has
been gained by combining mutant enzymes and analogs
[7,9,10]. The binding energy contributed by substrate
OH-groups has been determined for only a few carbohy-
drate active enzymes. Of these, the starch hydrolase
glucoamylase from Aspergillus niger has been the most
intensively examined [7,9±13].
Three-dimensional structures of protein±carbohydrate
complexes can guide and support protein engineering and
molecular recognition experiments. For family 13 glycoside
hydrolases, there are no crystal structures for a-glucosidases;
however, the structure of free Bacillus oligo-1,6-glucosidase
has been solved [14]. Furthermore, only a few a-glucosidases
are produced by heterologous gene expression, which is a
prerequisite for structure±function relationship investiga-
tions by site-directed mutagenesis [15±21]. While the yeast
genome is known and thus the primary structures of its
a-glucosidases, the sequenced strain of Saccharomyces
cerevisiae is not necessarily identical to the baker's yeast
used as a source of enzymes in the present study and
sequences have not been reported for brewer's yeast
enzymes. In view of this limited information, use of synthetic
substrate analogs is particularly attractive for gaining
knowledge of the nature and strength of substrate±
a-glucosidase interactions. Thus using deoxy-analogs key
polar groups in maltose were identi®ed for high pI barley
a-glucosidase of glycoside hydrolase family 31 to be OH-4¢
and -6¢with minor contributions for OH-3¢,-2¢,and-3
[13, 22].
Yeast a-glucosidase and oligo-1,6-glucosidase are exo-
acting glycoside hydrolases catalyzing release of a-
D
-glucose
from nonreducing ends of various a-linked substrates. The
enzymes are further subclassi®ed into type I, hydrolysing
heterogeneous substrates like aryl glucosides and sucrose
more ef®ciently than maltose; type II being highly active on
maltose and isomaltose but of low activity toward aryl
glucosides; and type III resembling type II, but hydrolysing
Correspondence to B. Svensson, Department of Chemistry, Carlsberg
Laboratory, DK-2500 Copenhagen Valby, Denmark;
Fax: + 45 33 27 47 08; Tel.: + 45 33 27 53 45;
E-mail: bis@crc.dk
Enzymes:a-glucosidase (a-
D
-glucoside glucohydrolase, EC 3.2.1.20);
oligo-1,6-glucosidase (dextrin 6-a-glucanohydrolase, EC 3.2.1.10);
glucoamylase (a-
D
-glucan glucohydrolase, EC 3.2.1.3).
*Present address: Pantheco, Bùge Alle
Â, DK 2970 Hùrsholm Denmark.
Dedication: this paper is dedicated to Prof. Joachim Thiem on the
occasion of his 60
th
birthday.
(Received 12 October 2001, revised 26 November 2001, accepted 30
November 2001)
Eur. J. Biochem. 269, 728±734 (2002) ÓFEBS 2002

di- and oligo-saccharides and starch at comparable rates
[23,24]. The sequence classi®es a-glucosidases in glycoside
hydrolase families 13 and 31 [25±27]. Yeast a-glucosidases
and oligo-1,6-glucosidase belong to family 13 and are
of type I that prefers p-nitrophenyl-a-
D
-glucopyranoside
[28].
Glycoside hydrolase family 13 (or Ôthe a-amylase familyÕ)
currently comprises 28 speci®cities of amylolytic and related
enzymes. Several crystal structures of enzyme-inhibitor
complexes highlight active sites created by b®asegments
in catalytic (b/a)
8
barrel domains (reviewed in [29±31]).
Because no ligand complex is available of oligo-1,6-
glucosidase, the only structure-determined exo-acting
a-glucosidase [14], side-chains participating in substrate
binding and catalysis are solely identi®ed by sequence
comparison. Clearly a-glucosidases lack the sequence motif
in b®aloop 4 of family 13 [30] containing residues
binding substrate at subsite +2 (nomenclature as in [32]) in
a-amylases, cyclodextrin glycosyltransferases, and related
enzymes [30,33±37].
In this study, seven monodeoxygenated isomaltosides are
used to map substrate OH-groups required by yeast
a-glucosidases and oligo-1,6-glucosidase in hydrolysis of
the a-1,6-glucosidic bond. The energy contributed by each
OH-group for transition-state stabilization re¯ects the
strength of a speci®c protein±carbohydrate contact and
energy pro®les for the a-glucosidases and oligo-1,6-glucosi-
dase are compared.
1
H-NMR spectroscopy was used to
con®rm that all enzymes hydrolyse isomaltose with reten-
tion of anomeric con®guration characteristic of family 13
(reviewedin[38]).
The exo-acting glucoamylase similarly to the a-glucosid-
ases catalyses the releases of glucose from the nonreducing
ends of substrates, but with inversion of the anomeric
con®guration [39]. While glucoamylase prefers the R-isomer
of isomaltose diastereoisomeric analogs [40], a-glucosidase
in the present study selects methyl 6-S-ethyl-a-isomaltoside
in preference to the R-isomer. Molecular recognition of
isomaltosides is more similar for the yeast a-glucosidase and
oligo-1,6-glucosidase of glycoside of hydrolase family 13
when compared to that of glucoamylase of glycoside
hydrolase family 15 or of a type II a-glucosidase from the
retaining glycoside hydrolase family 31 [9,10,40,41].
MATERIALS AND METHODS
Enzymes and substrates
Oligo-1,6-glucosidase from baker's yeast (EC 3.2.1.10;
Lot no. 23H8080), and a-glucosidases from brewer's
(EC 3.2.1.20; Type VI; Lot no. 21F8105) and baker's
(EC 3.2.1.20; Type I; Lot no. 122H8000) yeast were
obtained from Sigma. After dissolution in 50 m
M
phosphate
pH 6.8 (a-glucosidases) or 50 m
M
sodium maleate pH 6.8
(oligo-1,6-glucosidase) followed by extensive dialysis at 4 °C
against these buffers, the different enzymes (oligo-1,6-
glucosidase, 30 UámL
)1
; brewer's yeast a-glucosidase,
200 UámL
)1
; baker's yeast a-glucosidase, 61 UámL
)1
)were
used without further puri®cation in the kinetic and stereo-
chemical studies. One unit is de®ned as the amount of
enzyme required to liberate 1 lmol of glucose from
p-nitrophenyl a-
D
-glucoside (Sigma) per min at 30 °C. The
synthesized methyl a-isomaltoside, seven monodeoxy-
genated methyl a-isomaltosides [42], methyl 6-R-C-ethyl-
and methyl 6-S-C-ethyl-a-isomaltoside [41] were generous
gifts of U. Spohr and the late R. U. Lemieux, University of
Alberta, Edmonton, Canada.
Enzyme assays
a-Glucosidase activity was determined at 30 °Cin0.1
M
sodium maleate, pH 6.8 (oligo-1,6-glucosidase) or 50 m
M
phosphate, pH 6.8 (a-glucosidases). Glucose [7,10,40,41]
was analysed for analogs at the reducing end ring (reaction
volume 100 lL), aliquots (15 lL) being transferred at
regular time intervals to microtiter plate wells already
containing quench solution (200 lL1
M
Tris, pH 7.6,
5UámL
)1
glucose oxidase (A. niger), 1 UámL
)1
peroxidase
(horseradish), and 0.21 mgámL
)1
o-dianisidine). Absor-
bances were read at 450 nm after 1 h incubation at
room temperature using a microtiter plate reader (Ceres
UV900Hdi, Bio-Tek), and quanti®ed using
D
-glucose as a
standard [22,40]. Deoxygenated glucose analogs were ana-
lysed essentially as described [10,40,41] with substrate
analogs at the nonreducing end sugar (reaction volume
400 lL) aliquots (100 lL) were transferred to quench buffer
containing 60 UámL
)1
glucose oxidase, 1 UámL
)1
peroxi-
dase, and 0.1 mgámL
)1
o-dianisidine, and the absorbances
were read at 450 nm after 4 h incubation at room
temperature, and quanti®ed using the relevant deoxygenated
D
-glucose as standard. The a-glucosidase catalyzed hydro-
lysis was initiated by addition of 0.1±91 U enzyme. The
limited amounts of deoxygenated analogs available allowed
only determination of second-order rate constants, V
max
/K
m
(s
)1
áU
)1
)v
o
/E
o
S
o
,wherev
o
is the initial rate of hydro-
lysis, S
o
the initial substrate concentration, and E
o
the
amount of enzyme in U. Two S
o
concentrations of around
0.1 ´K
m
were used to ensure that substrate hydrolysis was
linear with time. The increase in activation energy due
to substrate deoxygenation was calculated by DDGà
±RTln[(V
max
/K
m
)
a
/(V
max
/K
m
)
b
][43],whereareferstoana-
log and b to parent substrate. For the two diastereoisomers,
V
max
and K
m
were determined by ®tting initial rates at eight
different substrate concentrations from 0.1 ´K
m
to 4 ´K
m
to the Michealis±Menten equation essentially as described
previously [40].
Reaction stereochemistry
Lyophilized enzymes were redissolved in 0.1
M
sodium
phosphate pH 6.8 in D
2
O and the stereochemistry of
isomaltose hydrolysis was determined by
1
HNMRat
310 K using a Bruker AMX-600 spectrometer operated at
600 MHz. After recording the substrate spectrum of
100 m
M
isomaltose (in 600 lL0.1
M
phosphate, pH 6.8,
in D
2
O), enzyme was added (oligo-1,6-glucosidase, 40 U;
baker's, 135 U and brewer's yeast a-glucosidase, 140 U)
and reactions monitored by recording spectra at regular
intervals.
RESULTS AND DISCUSSION
Energetics of deoxy isomaltoside hydrolysis
V
max
/K
m
values for hydrolysis of methyl a-isomaltoside are
comparable for the three enzymes, the a-glucosidases from
ÓFEBS 2002 Substrate recognition in yeast a-glucosidases (Eur. J. Biochem. 269) 729
brewer's and baker's yeast showing 31 and 164% of the
activity of the oligo-1,6-glucosidase, respectively (Table 1).
Furthermore, the activity of the three enzymes was reduced
by roughly the same extent, i.e. 440±3400-, 560±2900-, and
1350±8800-fold by substrate deoxygenation at OH-2¢,-3¢,
-4¢,or-6¢(Table 1). The losses in activity compared to the
parent substrate for all enzymes were smallest for the
6¢-deoxy analog and largest for the 2¢-deoxy analog, while
intermediary losses in activity for 3¢-and4¢-deoxy analogs
did show small variations among the enzymes (Table 1).
For the two a-glucosidases, deoxygenation at the reducing
end ring of the substrate had no effect or a very minor effect,
the activity varying relative to the parent substrate by
factors of 0.84±1.4 and 0.42±1.0 for the enzymes from
brewer's and baker's yeast, respectively. In contrast, for
oligo-1,6-glucosidase, the deoxy-2, -3, and -4 analogs
showed ninefold, ®vefold, and no reduction in V
max
/K
m
,
respectively (Table 1).
The DDGàcalculated from the V
max
/K
m
values deter-
mined for a given analog and the parent substrate,
respectively, indicated the energy contributed to transition-
state stabilization by corresponding the OH-group. Because
DDGàfor the four deoxy-analogs at the nonreducing sugar
ring, that binds to the enzymes at subsite )1, was in the
range 16.1±24.0 kJámol
)1
for the three enzymes (Table 1),
the removal of one of the OH-groups from this ring
dramatically affected substrate hydrolysis. These
OH-groups can therefore be considered key polar groups
and most likely interact with charged residues on the
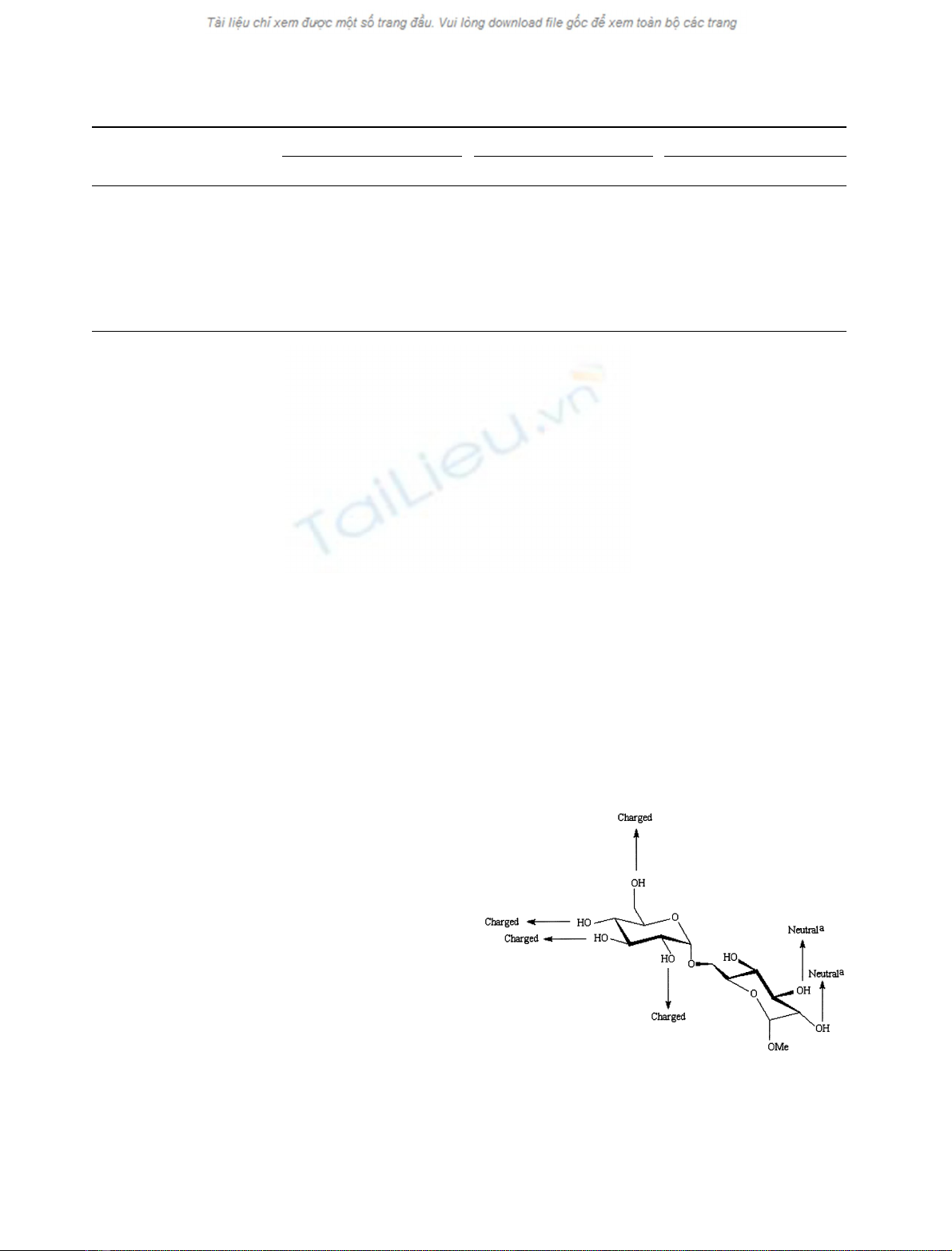
proteins [44] (Fig. 1). At the reducing end ring, however,
DDGàvalues of 4±6 kJámol
)1
for oligo-1,6-glucosidase
(Table 1) were obtained by replacement of the OH-2
and -3 groups, respectively, suggesting that these
OH-groups participate in neutral hydrogen bonds with the
enzyme (Fig. 1). The OH-4 did not seem important in
substrate binding and hydrolysis.
The three-dimensional structure of oligo-1,6-glucosidase
from Bacillus cereus [14], is currently the only available
structure of any type of a-glucosidases. This enzyme has an
N-terminal (b/a)
8
barrel common to glycoside hydrolase
family 13 [30,31], a domain B that protrudes from the barrel
bstrand 3, and a C-terminal Greek key motif. Moreover
several extra-barrel secondary structure elements occur in
the segments that connect the bstrands to the ahelices of the
(b/a)
8
barrel fold [14]. The catalytic site is located at the
bottom of a cleft between domain B and several of the
b®aconnecting segments [14,30]. The molecular recog-
nition of isomaltose analogs described above indicate very
strong interaction of the nonreducing substrate ring at the
enzyme subsite )1, most probably with charged side chains,
as a major driving force for stabilization of the enzyme±
substrate transition-state. Several of the side-chains inter-
acting at subsite )1 will belong to the consensus sequence
motifs containing catalytic acids, transition-state stabilizing
histidines, and structurally important arginine and aspartate
side chains [30].
While protein±substrate contacts at subsite )1provide
major binding energy, the distribution and strength of
intermolecular hydrogen bonds involving the aglycon
moiety and subsite +1, as well as subsites beyond subsite
+1 in type III a-glucosidases, exhibit substrate speci®city
variation among the a-glucosidases. The yeast a-glucosid-
ases as reported here only show protein±carbohydrate
hydrogen bonding involving subsite )1,andnosugar
OH-groups associated stabilization energy was critical for
accommodation at subsite +1. As shown in Table 2, these
a-glucosidases that do not require hydrogen bonding to the
Table 1. Speci®city constants and DDGà
a
(kJámol
)1
)fora-glucosidase catalyzed hydrolysis of methyl a-isomaltoside and a series of mono-deoxy-
genated analogs.
Oligo-1,6-glucosidase
b
a-glucosidase (brewer's yeast)
c
a-Glucosidase (baker's yeast)
d
V
max
/K
m
(s
)1
áU
)1
)DDGàV
max
/K
m
(s
)1
áU
)1
)DDGàV
max
/K
m
(s
)1
áU
)1
)DDGà
Methyl-a-isomaltoside 1.4 ´10
)4
0.8 ´10
)5e
± 4.4 ´10
)5
2.9 ´10
)6
± 2.3 ´10
)4
3.2 ´10
)6
±
2-Deoxy-methyl-a-isomaltoside 1.6 ´10
)5
0.9 ´10
)6
5.8 3.7 ´10
)5
4.9 ´10
)6
0.5 9.7 ´10
)5
5.8 ´10
)6
2.3
3-Deoxy-methyl-a-isomaltoside 2.9 ´10
)5
0.6 ´10
)6
4.1 6.0 ´10
)5
1.4 ´10
)5
)0.8 1.3 ´10
)4
1.3 ´10
)5
15
4-Deoxy-methyl-a-isomaltoside 1.4 ´10
)4
1.1 ´10
)5
± 5.7 ´10
)5
0.8 ´10
)6
)0.7 2.4 ´10
)4
2.0 ´10
)6
)0.1
2¢-Deoxy-methyl-a-isomaltoside 4.1 ´10
)8
5.5 ´10
)9
21.5 1.5 ´10
)8
1.2 ´10
)9
21.2 2.6 ´10
)8
3.0 ´10
)9
24.0
3¢-Deoxy-methyl-a-isomaltoside 1.1 ´10
)7
6.5 ´10
)9
18.9 2.7 ´10
)8
5.3 ´10
)9
19.6 2.7 ´10
)8
3.1 ´10
)9
23.9
4¢-Deoxy-methyl-a-isomaltoside 1.0 ´10
)7
1.1 ´10
)8
19.1 1.5 ´10
)8
4.4 ´10
)10
21.2 3.2 ´10
)8
2.8 ´10
)9
23.5
6¢-Deoxy-methyl-a-isomaltoside 3.2 ´10
)7
2.1 ´10
)8
16.1 7.9 ´10
)8
4.7 ´10
)9
16.7 1.4 ´10
)7
1.5 ´10
)8
19.6
a
DDGà)RT ln[(V
max
/K
m
)
a
/(V
max
/K
m
)
b
] [43], where a and b refer to analog and parent substrate, respectively;
b
At 30 °C using 0.1
M
sodium maleate, pH 6.8;
c
At 30 °C using 50 m
M
phosphate, pH 6.;
d
At 30 °C using 50 m
M
phosphate, pH 6.8;
e
standard deviation.
Fig. 1. Schematic representation of proposed intermolecular hydrogen
bond interactions between isomaltose and a-glucosidases from baker's
and brewer's yeast and from oligo-1,6-glucosidase from baker's yeast.
a
, only for oligo-1,6-glucosidase. Invariant glycoside hydrolase family
13 side chain candidates of interaction with the four nonreducing
substrate ring OH-groups are described in detail in a recent review [30].
730 T. P. Frandsen et al. (Eur. J. Biochem. 269)ÓFEBS 2002


























%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)