
Lid L11 of the glutamine amidotransferase domain
of CTP synthase mediates allosteric GTP activation
of glutaminase activity
Martin Willemoe
¨s
1
, Anne Mølgaard
1,2
, Eva Johansson
1,3
and Jan Martinussen
4
1 Centre for Crystallographic Studies, Department of Chemistry, University of Copenhagen, Denmark
2 Center for Biological Sequence Analysis, BioCentrum-DTU, The Technical University of Denmark, Lyngby, Denmark
3 European Synchrotron Radiation Facility, Grenoble Cedex, France
4 Microbial Physiology and Genetics, BioCentrum-DTU, The Technical University of Denmark, Lyngby, Denmark
CTP synthase (EC 6.3.4.2) catalyses the synthesis of
CTP by amination of the 4-position of the pyrimidine
moiety of UTP [1]. The enzyme is a homotetramer,
each subunit consisting of two domains; the
N-terminal synthase domain where CTP formation
takes place and the C-terminal GATase domain
Keywords
allosteric regulation; flexible loop;
Lactococcus lactis; nucleotide metabolism;
oxy-anion hole
Correspondence
M. Willemoe
¨s, Centre for Crystallographic
Studies, Department of Chemistry,
University of Copenhagen,
Universitetsparken 5, Copenhagen
DK-2100 Ø, Denmark
Fax: +45 3532 0299
Tel: +45 3532 0239
E-mail: martin@ccs.ki.ku.dk
(Received 25 November 2004, revised 8
December 2004, accepted 10 December
2004)
doi:10.1111/j.1742-4658.2004.04525.x
GTP is an allosteric activator of CTP synthase and acts to increase the k
cat
for the glutamine-dependent CTP synthesis reaction. GTP is suggested, in
part, to optimally orient the oxy-anion hole for hydrolysis of glutamine that
takes place in the glutamine amidotransferase class I (GATase) domain of
CTP synthase. In the GATase domain of the recently published structures
of the Escherichia coli and Thermus thermophilus CTP synthases a loop
region immediately proceeding amino acid residues forming the oxy-anion
hole and named lid L11 is shown for the latter enzyme to be flexible and
change position depending on the presence or absence of glutamine in the
glutamine binding site. Displacement or rearrangement of this loop may
provide a means for the suggested role of allosteric activation by GTP to
optimize the oxy-anion hole for glutamine hydrolysis. Arg359, Gly360 and
Glu362 of the Lactococcus lactis enzyme are highly conserved residues in lid
L11 and we have analyzed their possible role in GTP activation. Characteri-
zation of the mutant enzymes R359M, R359P, G360A and G360P indicated
that both Arg359 and Gly360 are involved in the allosteric response to GTP
binding whereas the E362Q enzyme behaved like wild-type enzyme. Apart
from the G360A enzyme, the results from kinetic analysis of the enzymes
altered at position 359 and 360 showed a 10- to 50-fold decrease in GTP
activation of glutamine dependent CTP synthesis and concomitant four- to
10-fold increases in K
A
for GTP. The R359M, R359P and G360P also
showed no GTP activation of the uncoupled glutaminase reaction whereas
the G360A enzyme was about twofold more active than wild-type enzyme.
The elevated K
A
for GTP and reduced GTP activation of CTP synthesis of
the mutant enzymes are in agreement with a predicted interaction of bound
GTP with lid L11 and indicate that the GTP activation of glutamine
dependent CTP synthesis may be explained by structural rearrangements
around the oxy-anion hole of the GATase domain.
Abbreviations
ATPcS, adenosine 5¢-[c-thio]triphosphate; GATase, class I glutamine amidotransferase; PDB ID, Protein Data Bank entry.
856 FEBS Journal 272 (2005) 856–864 ª2005 FEBS

responsible for the hydrolysis of glutamine [2]. In addi-
tion the enzyme has a site for GTP that acts as an
allosteric activator. The reaction proceeds via the
intermediate 4-phosphoryl UTP generated by ATP-
dependent phosphorylation [3,4]. The amino group
transferred to this activated intermediate is either
obtained from glutamine hydrolysis or ammonia pre-
sent in the solution [1]. The rate of the glutamine
dependent CTP synthesis reaction is greatly stimulated
by the allosteric binding of GTP and this activation
has been the focus of several reports [5–9]. CTP syn-
thase is the only member of the family of GATase
domain harboring enzymes where an allosteric effector
regulates the glutaminase activity. However, only
recently has it been possible functionally to associate
individual amino acid residues, Thr-431 and Arg433 in
Lactococcus lactis CTP synthase [10] and Arg429 in
the Escherichia coli enzyme [11], with this property of
the glutamine dependent CTP formation. For the
L. lactis enzyme, allosteric binding of GTP acts in syn-
ergy with the 4-phoshorylated UTP intermediate to
activate glutamine dependent CTP synthesis [12]. In
addition, GTP appears to promote channeling of NH
3
derived from glutamine hydrolysis to the synthase site
[9]. The nature of the formation of this channel and
the role GTP may play in forming this, has recently
been suggested from analysis of the crystal structure of
the E. coli enzyme. Based on structural homology to
GTP binding enzymes, GTP was modeled into the
apo-structure of CTP synthase and was suggested to
bind in a cleft between the GATase domain and the
synthase domain [13] (Fig. 1A). Recently, an E. coli
mutant enzyme altered at position 109 (L109A) that is
impaired in the coupling between ammonia derived
from glutamine hydrolysis and CTP formation due to
a constricted or leaky ammonia tunnel [14], has provi-
ded evidence for this putative binding site for GTP
and the role GTP plays in coupling of glutamine
hydrolysis and CTP synthesis [13].
In a work on inhibition of the E. coli enzyme by
the analogue glutamate c-semialdehyde that mimics
intermediates of glutamine hydrolysis, Bearne and
coworkers [6] suggested that the oxy-anion hole is a
target for GTP regulation of the glutaminase activity
of CTP synthase. A comparison of part of the CTP
synthase GATase domain (Fig. 1B) with other known
structures of enzymes incorporating this catalytic
domain (Fig. 1C) showed that whereas the central
beta-sheet and the alpha helices superimpose well, the
loop regions differ, including the region that proceeds
from the oxy-anion hole into a common alpha helix.
This region with a variable loop structure has been
named lid L11 in the E. coli CTP synthase structure
[13] and we will use this nomenclature when referring
to this structural region in the following. The apo-
structure of the E. coli enzyme [13] and the structure
of the Thermus thermophilus CTP synthase [15] in a
complex with glutamine are virtually superimposable.
However, the apo-structure and the structure in com-
plex with sulfate ions of the T. thermophilus enzyme
deviates significantly in the Ca-chain of this region
(Fig. 1B). In fact some residues of the lid L11 were
not detected in the electron density maps [15] indica-
ting that this region is highly flexible and can adapt
different structures.
Even though there is currently no detailed structural
information that explains the activation of glutamine
hydrolysis in GATase domains upon binding of amino
acceptor substrates [16], it is likely to involve small
movements or re-organizations of the environment
around the oxy-anion hole [6,17–19]. The importance
of the loop corresponding to lid L11 has previously
been demonstrated for the E. coli carbamoyl phos-
phate synthase that has a cysteine residue (Cys248)
within this loop. When this residue is labeled with
N-ethylmaleimide [20] or changed to more bulky resi-
dues (Arg, Asp, Phe and Trp) [17], it results in
uncoupling of glutaminase activity from carbamoyl
phosphate synthesis. In the case of the C248D enzyme,
glutamine-dependent carbamoyl phosphate synthesis is
completely abolished and large increases in glutami-
nase activity and K
M
for glutamine are observed [17].
The dramatic effect on glutamine hydrolysis and coup-
ling of the reactions can be explained by local changes
in the structure of the region corresponding to lid
L11 [21].
Residues 354–357 of the L. lactis enzyme forms
part of the oxy-anion hole and homologues residues
are found with little variation in all known GATase
domains [22]. The corresponding residues from the
GATase domain of E. coli CTP synthase [23] were
previously subjected to mutational analysis. One
mutant enzyme where Gly352 was changed to a pro-
lyl (Gly355 in the L. lactis enzyme) had lost detect-
able glutamine-dependent CTP synthase activity and
could not be labeled by [
14
C]6-diazo-5-oxonorleucine,
but was still active with NH
4
Cl as a substrate. The
result from this study may be interpreted in terms of
the recent CTP synthase structure, as this residue is
positioned at a pivotal point at which the preceding
rigid part of the oxy-anion hole connects with the flexi-
ble lid L11 (Fig. 1B). Flexibility or displacement of lid
L11 may be a prerequisite for the activation of the
glutaminase activity hampered by a glycine to a proline
substitution at position 352 (355 in the L. lactis
enzyme).
M. Willemoe
¨set al. GTP activation of CTP synthase
FEBS Journal 272 (2005) 856–864 ª2005 FEBS 857
Taken together, the above observations all point to
the oxy-anion hole and its immediate surroundings as
a target for regulation of GATases. This prompted us
to investigate whether highly conserved amino acid
residues surrounding the oxy-anion hole plays a role in
GTP activation of glutamine hydrolysis by the L. lactis
CTP synthase. We chose to analyze by site-directed
mutagenesis the role of the highly conserved residues
Arg359, Gly360 and Glu362 (Fig. 1D), that are part in
lid L11. Arg359 was changed to methionyl to maintain
the hydrophobic part of the arginyl side chain while
deleting the guanidinium group. The Gly360 to alanyl
and Glu362 to glutaminyl substitutions are both
allowed for at these positions (Fig. 1D), but do reduce
the backbone flexibility or delete the charge of the side
chain, respectively. In addition, we replaced Arg359
and Gly360 with a prolyl to test the importance of the
flexibility of lid L11 based on the assumption that a
prolyl would restrict the flexibility of the Ca-chain at
these positions.
AB
CD
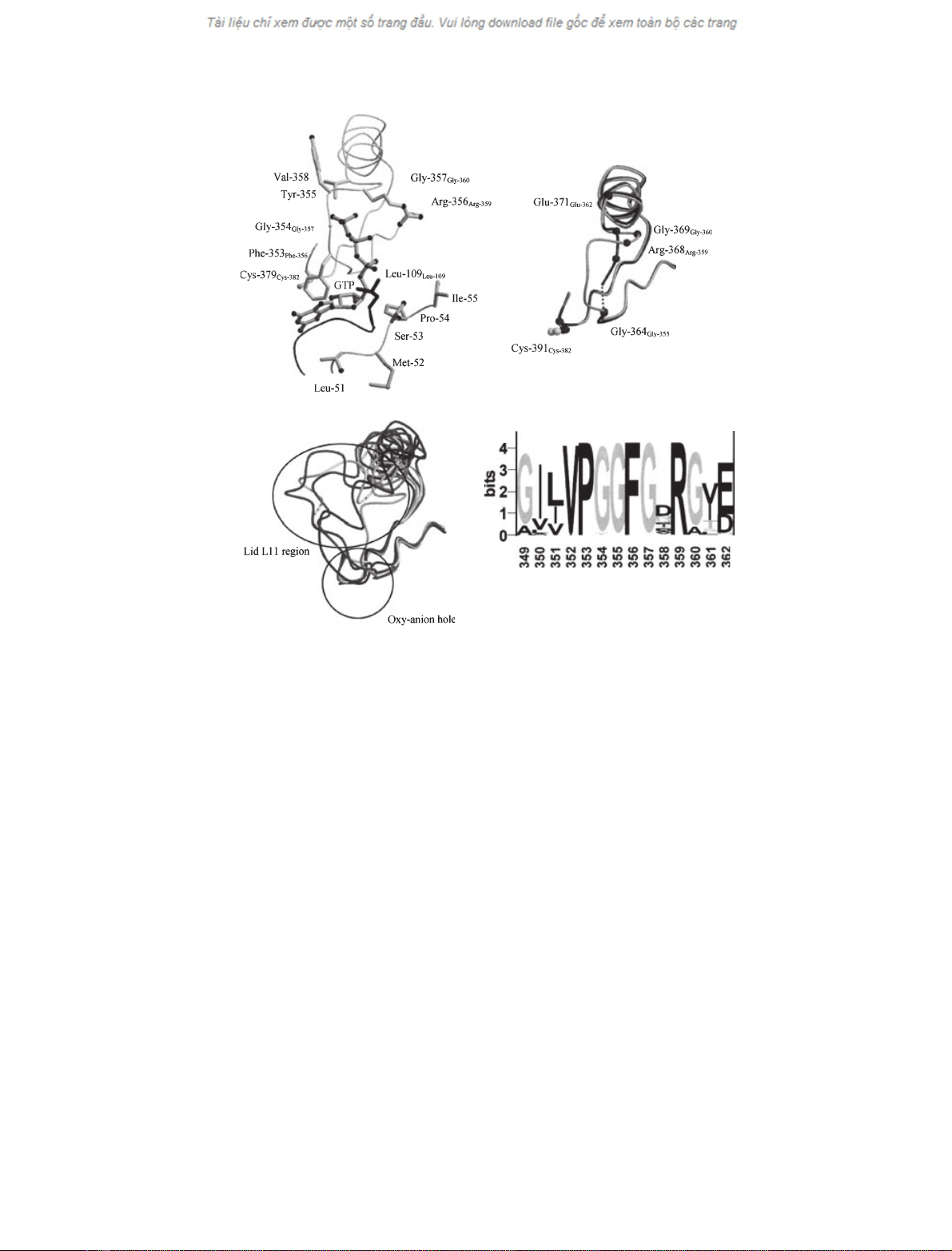
Fig. 1. The structure and role of lid L11 in CTP synthase. (A) The proposed binding of GTP modelled into the apo-structure of the E. coli CTP
synthase by Endrizzi et al. [13]. The residues are numbered according to the E. coli enzyme while numbers in subscript are according to
the L. lactis sequence. Shown are: residues 346–380 including the catalytic Cys379 of the GATase domain, residues 51–55 and residues
104–110 of the synthase domain. (B) Comparison of the structures for T. thermophilus CTP synthase (residues 358–392) in complex with
glutamine (PDB ID; 1VCO, light grey) or sulfate (PDB ID; 1VCN, dark grey). In the latter structure residues 365 and 366 (corresponding to
356 and 357 in the L. lactis enzyme) are missing from the structure as indicated by the dotted line. The residues of lid L11 are numbered
according to the T. thermophilus sequence while numbers in subscript are according to the L. lactis sequence. The side chain of the catalytic
Cys391 is shown for orientation. (C) Comparison of the oxy-anion hole and surroundings of GATase domains from CTP synthase (residues
346–380, PDP ID; 1S1M), carbamoyl phosphate synthase small domain (residues 235–270, PDB ID; 1BXR), GMP synthase (residues 53–87,
PDB ID; 1GPM) and anthranilate synthase (residues 48–85, PDB ID; 1QDL) and imidazole glycerol phosphate synthase (residues 47–87, PDB
ID; 1JVN).The oxy-anion hole and the loops corresponding to L11 in CTP synthase are encircled. (A), (B), and (C) were prepared with
MOLSCRIPT [30] and RASTER3D[31]. (D) A sequence logo [32] was generated of the sequence region of interest (L. lactis CTP synthase num-
bering), based on a BLAST [33] alignment of 43 full-length sequences annotated as CTP synthase. The height of each residue is proportional
to its frequency, and the most common residue is on top at each position. The total height of each position is adjusted to signify the extent
of conservation at that position [34].
GTP activation of CTP synthase M. Willemoe
¨set al.
858 FEBS Journal 272 (2005) 856–864 ª2005 FEBS

























