
N-Glycosylation is important for the correct intracellular
localization of HFE and its ability to decrease cell surface
transferrin binding
Lavinia Bhatt
1
, Claire Murphy
2
, Liam S.O’Driscoll
2
, Maria Carmo-Fonseca
3
, Mary W. McCaffrey
1
and John V. Fleming
2,3
1 Department of Biochemistry, Biosciences Institute, University College Cork, Ireland
2 Department of Biochemistry, School of Pharmacy and ABCRF, University College Cork, Ireland
3 Institute of Molecular Medicine, University of Lisbon, Portugal
Keywords
HFE; N-glycosylation; transferrin; transferrin
receptor 1; b2-microglobulin
Correspondence
J. V. Fleming, Department of Biochemistry
and School of Pharmacy, University College
Cork, Cork, Ireland
Fax: +353 21 4901656
Tel: +353 21 4901679
E-mail: j.fleming@ucc.ie
Note
L. Bhatt and C. Murphy contributed equally
to this work
(Received 8 February 2010, revised 14 May
2010, accepted 2 June 2010)
doi:10.1111/j.1742-4658.2010.07727.x
HFE is a type 1 transmembrane protein that becomes N-glycosylated dur-
ing transport to the cell membrane. It influences cellular iron concentra-
tions through multiple mechanisms, including regulation of transferrin
binding to transferrin receptors. The importance of glycosylation in HFE
localization and function has not yet been studied. Here we employed bio-
informatics to identify putative N-glycosylation sites at residues N110,
N130 and N234 of the human HFE protein, and used site-directed muta-
genesis to create combinations of single, double or triple mutants. Com-
pared with the wild-type protein, which co-localizes with the type 1
transferrin receptor in the endosomal recycling compartment and on dis-
tributed punctae, the triple mutant co-localized with BiP in the endoplas-
mic reticulum. This was similar to the localization pattern described
previously for the misfolding HFE-C282Y mutant that causes type 1 hered-
itary haemachromatosis. We also observed that the triple mutant was func-
tionally deficient in b2-microglobulin interactions and incapable of
regulating transferrin binding, once again, reminiscent of the HFE-C282Y
variant. Single and double mutants that undergo limited glycosylation
appeared to have a mixed phenotype, with characteristics primarily of the
wild-type, but also some from the glycosylation-deficient protein. There-
fore, although they displayed an endosomal recycling compartment/punc-
tate localization like the wild-type protein, many cells simultaneously
displayed additional reticular localization. Furthermore, although the
majority of cells expressing these single and double mutants showed
decreased surface binding of transferrin, a number appeared to have lost
this ability. We conclude that glycosylation is important for the normal
intracellular trafficking and functional activity of HFE.
Structured digital abstract
lMINT-7896236,MINT-7896218:beta2M (uniprotkb:P61769)physically interacts (MI:0915)
with HFE (uniprotkb:Q30201)byanti bait coimmunoprecipitation (MI:0006)
lMINT-7896162:TfR1 (uniprotkb:P02786) and HFE (uniprotkb:Q30201)colocalize (MI:0403)
by fluorescence microscopy (MI:0416)
Abbreviations
ER, endoplasmic reticulum; ERC, endosomal recycling compartment; HH, hereditary haemachromatosis; b2M, b2 microglobulin; MHC, major
histocompatability complex; PNGase F, N-glycosidase F; Tfn, transferrin; TfR1, transferrin receptor 1; TfR2, transferrin receptor 2.
FEBS Journal 277 (2010) 3219–3234 ª2010 The Authors Journal compilation ª2010 FEBS 3219

Introduction
The hereditary haemochromatosis (HH) protein HFE
(high Fe) is a type 1 transmembrane protein that plays
an important role in controlling physiological iron
homeostasis [1–3]. It is widely expressed throughout
the body with expression highest in cells that are
involved in iron metabolism [4–6]. Mutations in the
HFE protein cause type 1 HH, which is an inherited
disease of iron metabolism that results in iron overload
in several organs [4,7]. The HFE mutation detected in
the majority of HH patients results in the replacement
of cysteine residue 282 with tyrosine (C282Y). The
mutant protein is unable to form a structurally impor-
tant disulfide bridge required for HFE interactions
with b2 microglobulin (b2M) [4,5,8–11]. In the absence
of b2M binding, the protein misfolds and is retained in
the endosplasmic reticulum (ER) where it induces an
unfolded protein stress response that is characterized
by alternative splicing of XBP-1 and increased expres-
sion of CHOP and BiP [12–14]. A second well-
described HFE mutation associated with HH leads to
the replacement of histidine at residue 63 with aspar-
tate. This mutant is capable of b2M interaction and
cell-surface expression but is unable to regulate cellular
iron uptake like the wild-type HFE protein [4,15].
Although much insight into HFE function has been
gained through studying the cellular and biochemical
properties of these different mutant proteins, the exact
mechanism by which HFE regulates intracellular iron
levels is still not completely understood.
The HFE primary sequence exhibits significant
homology to major histocompatability complex
(MHC) class I molecules and the protein is organized
into a1, a2 and a3 structural domains that resemble
those described for MHC class I and related proteins
[4,16]. The N-terminal a1 and a2 domains come
together to form a superstructure composed of two
ahelices layered on top of eight anti-parallel bsheets.
In MHC class I proteins this a1/a2 superstructure
forms a peptide-binding groove that mediates antigen
binding and presentation to CD8
+
cytolytic T cells.
In HFE, the proximity of the two ahelices and the
presence of amino acid side chains that project into
the groove appear to prevent peptide binding [16]. The
a3 region, like its homologous domain in MHC
class I, is an immunoglobulin-like domain that medi-
ates binding to b2M [16,17]. C-Terminal residues of
HFE mediate its retention in the cell membrane.
Shortly after HFE was discovered it was reported to
co-localize and interact with the type 1 transferrin
receptor (TfR1) [5,18]. TfR1 mediates the endocytosis
of iron-loaded transferrin into acidic endosomes where
the iron is released and transported into the cytoplasm
via the Nramp2-DCT1 iron transporter. Apo-transfer-
rin and TfR1 are recycled to the cell surface where
apo-transferrin is released [3,19]. Crystallography stud-
ies suggest that the a3 stem of HFE lies parallel to the
cell membrane and that the a1/a2 superstructure inter-
acts with helical regions located within TfR1. In this
way, it is possible for two HFE proteins to be posi-
tioned at either side of the TfR1 homodimer and form
a tetrameric complex that exhibits twofold symmetry
[17]. Reports from crystallography experiments have
been supported by mutagenesis studies that identified
residues located at the end of an a-helical region of the
HFE a1 domain (V100 and W103A) as being of par-
ticular importance for TfR1 interactions [16,17,20].
The effect of HFE binding to TfR1 is to lower the
affinity of the receptor for transferrin [15]. This most
likely reflects the existence of overlapping HFE and
transferrin-binding sites on the receptor [21,22]. Suc-
cessive studies indicate that HFE and TfR1 co-localize
during endosomal trafficking, although there are con-
tradictory reports as to whether TfR1 recycling is
affected by HFE [23–29].
Despite these well-described interactions, there is
mounting evidence that HFE regulation of cellular
iron levels may not depend solely on TfR1 binding
[30,31]. Attention has shifted to a second transferrin
receptor, TfR2, whose pattern of expression is more
restricted than that of ubiquitously expressed TfR1
[32]. Levels of TfR2 are highest in hepatocytes, the
predominant site of HFE expression, and recent stud-
ies have confirmed that the two proteins are capable of
interacting [33,34]. The nature of these interactions dif-
fers from those observed between HFE and TfR1 in
that they are mediated by the a3 domain of HFE, as
opposed to the a1/a2 superstructure [33]. An emerging
model, therefore, is that TfR2 competes with TfR1 for
lMINT-7896258,MINT-7896317,MINT-7896330,MINT-7896348,MINT-7896366:HFE (uni
protkb:Q30201) and transferrin (uniprotkb:P02787)colocalize (MI:0403)byfluorescence
microscopy (MI:0416)
lMINT-7896149:HFE (uniprotkb:Q30201) and BiP (uniprotkb:P11021)colocalize (MI:0403)
by fluorescence microscopy (MI:0416)
N-Glycosylation of HFE L. Bhatt et al.
3220 FEBS Journal 277 (2010) 3219–3234 ª2010 The Authors Journal compilation ª2010 FEBS

HFE binding. This occurs maximally at high concen-
trations of transferrin. The resulting HFE–TfR2 com-
plex, which is stabilized at high iron concentrations, is
believed to somehow regulate the expression of other
genes involved in iron metabolism. This includes hepci-
din, a 25 amino acid antimicrobial peptide that is
expressed in liver cells and is now recognized as a key
regulator of iron homeostasis in the body. Hepatocel-
lular hepcidin mRNA levels have been shown to be
regulated by HFE, and are altered in haemachromato-
sis patients with the C282Y mutation [35–37].
The importance of N-glycosylation with respect to
protein expression and function is highly variable.
Roles have been described in the secretion, stability
and oligomerization of proteins [38,39], the bioactivi-
ties of enzymes [40] and the binding affinities of
ligands and receptors [41]. In many instances, specific
functions can be attributed to glycosylation at specific
sites. For example, the human gonadotropin asubunit
has N-glycosylation sites at residues Asn52 and Asn78
that have been shown to differentially regulate receptor
signalling and secretion, respectively [38,39]. Another
example is the type 1 transferrin receptor, which has
N-glycosylation sites at residues Asn251, Asn317 and
Asn727. Mutation of Asn727 decreases cell-surface
expression, whereas mutation at the other two sites
does not [42].
HFE becomes glycosylated during post-translational
processing. Transfection studies have confirmed that
this involves N-glycosylation, and incubation of lysates
from HFE-expressing cells with N-glycosidase F
(PNGase F) leads to the accumulation of lower molec-
ular mass HFE proteins [13,43]. The carbohydrate
moiety undergoes processing and endoglycosi-
dase H-resistant HFE isoforms can be detected by
30 min post translation [10,13,18,23]. Although these
studies demonstrate that HFE is glycosylated, the
specific role, if any, that glycosylation might play in
cellular HFE function has not previously been studied.
In this article, we map the sites of HFE N-glycosyla-
tion and examine the importance of glycosylation on
parameters of protein localization and function.
Results
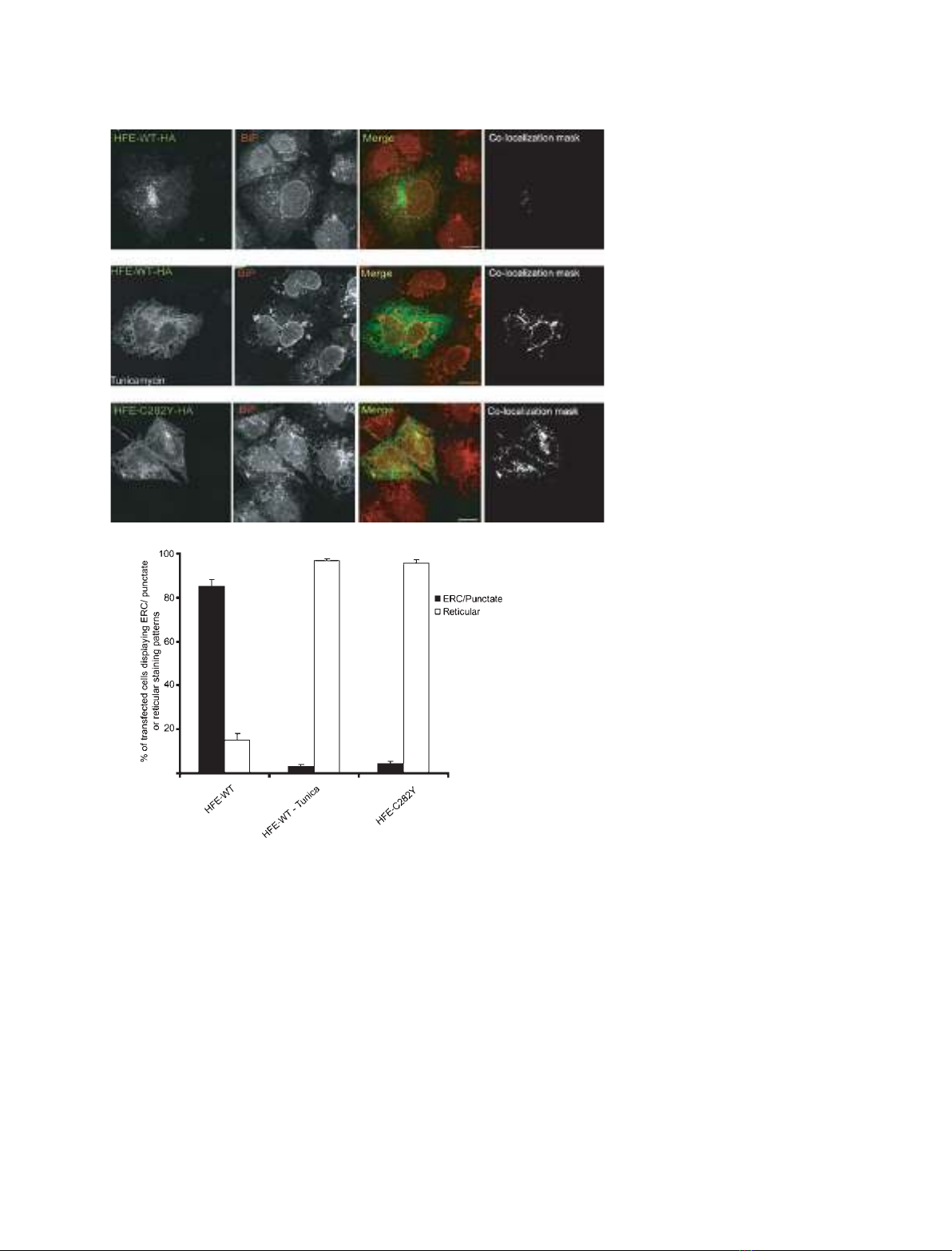
Tunicamycin treatment results in a reticular
pattern of HFE localization
Previous studies have demonstrated that HFE under-
goes post-translational N-glycosylation. As a first step
towards assessing the importance of N-glycosylation
on HFE expression, we transiently transfected HuTu80
to express HFE-WT–HA and cultured the cells in the
presence or absence of tunicamycin to inhibit glycan
production. Control, untreated cells predominantly
exhibited a punctate pattern of HFE expression with a
tubulovesicular concentration in the pericentrosomal
region (Fig. 1A,D), consistent with previous observa-
tions [11,28]. Immunostaining with anti-TfR1, anti-
Rab11a and Rab11-FIP3 Ig has identified the HFE-
containing pericentrosomal compartment of HuTu80
cells as the endosomal recycling compartment (ERC)
[28]. Treatment of HFE-WT–HA-expressing cells with
tunicamycin altered this pattern of localization and
resulted in a reticular pattern of cell localization
(Fig. 1B,D). Immunostaining showed significant
co-localization with the ER chaperone protein BiP,
demonstrating that the HFE-WT–HA was now locali-
zing primarily to the ER (Fig. 1B,D).
A similar pattern of reticular expression and BiP
co-localization was observed when HuTu80 cells were
transfected to express the HH-causing HFE-C282Y
variant (Fig. 1C,D), which has been shown through
multiple biochemical and microscopy approaches to be
retained in the ER [10,13,18,28,29].
HFE is glycosylated at residues Asn110, Asn130
and Asn234
Although the results in Fig. 1 point towards an impor-
tant role for glycosylation in HFE localization, it
remains possible that the effects of tunicamycin treat-
ment were indirect. To directly examine the impor-
tance of glycosylation on HFE, it was necessary to
generate an N-glycosylation-deficient mutant. To this
end, we used a bioinformatic prediction program
(netnglyc 1.0 Server; Technical University of Den-
mark) to identify putative glycosylation sites in the
protein. Consistent with previous predictions [18], we
identified three high-probability sites: asparagines at
positions 110, 130 and 234. Starting with wild-type
HFE, we generated all possible combinations of single
and double putative N-glycosylation site mutants
using site-directed mutagenesis. The wild-type and
mutant expression constructs were transfected into
HEK293T cells and the lysates analysed by western
blotting.
The results from these experiments, which are shown
in Fig. 2A, indicate that the introduction of single ala-
nine mutations at N110, N130 and N234, respectively,
resulted in the production of HFE proteins that
migrated with increased mobility on SDS/PAGE com-
pared with the wild-type. This suggested that all three
sites in the wild-type proteins are capable of becoming
glycosylated. The decrease in apparent molecular mass
became even more pronounced for proteins containing
L. Bhatt et al. N-Glycosylation of HFE
FEBS Journal 277 (2010) 3219–3234 ª2010 The Authors Journal compilation ª2010 FEBS 3221
combinations of double mutants, which displayed a
lower apparent molecular mass than either wild-type
or single mutant forms of the protein.
Although immunoblot analysis demonstrated that
the single and double mutants had decreased mass
compared with the wild-type protein, they still
appeared to be of higher molecular mass than the
unglycosylated form of the wild-type HFE protein –
which was produced when wild-type-expressing cells
were treated with tunicamycin (Fig. 2A; WT-Tunica).
This suggested that both the single and double
mutants were still partially glycosylated. To test this,
we transfected HEK293T cells to express either the
wild-type or mutant proteins, and incubated the cells
in the presence or absence of tunicamycin. Drug
treatments resulted in the accumulation of forms of
the mutant proteins that were of lower apparent
molecular mass and of similar size to the unglycosy-
lated form of the wild-type protein (see Fig. 2B for
single mutants and Fig. 2C for double mutants). This
suggested that the mutant proteins do indeed still
undergo limited glycosylation. For the single
mutants, additional supporting evidence for the per-
sistence of N-linked glycans was obtained by PNG-
ase F digestions of immunoprecipitated proteins,
which then migrated with lower apparent molecular
mass compared with the undigested forms (results
not shown).
A
B
C
D
Fig. 1. Inhibition of N-glycosylation influ-
ences patterns of HFE intracellular localiza-
tion. HuTu80 cells were transfected with
constructs expressing the HFE-WT–HA
(A, B) and HFE-C282Y–HA (C) proteins,
HFE-WT-expressing cells were incubated for
1 h in the absence (A) or presence (1B) of
2lgÆmL
–1
tunicamycin (Tunica) as indicated.
Cells were immunostained with anti-HA and
anti-BiP Ig, and processed for fluorescence
microscopy. Co-localization masks were
created as described in Materials and meth-
ods, and represent areas with overlapping
green and red pixels converted to white.
Scale bar, 10 lm. Identical results were
obtained when cells were transfected with
constructs directed to express amino-tagged
GFP–HFE-WT and GFP–HFE-C282Y, and in
general we found that HA and GFP tags
could be interchanged without altering the
pattern of cell localization (data not shown).
Figures shown are representative of at least
three independent experiments. (D) Graph
showing the relative amounts of transfected
cells exhibiting punctate or reticular localiza-
tion of expressed HFE proteins (n= 3).
N-Glycosylation of HFE L. Bhatt et al.
3222 FEBS Journal 277 (2010) 3219–3234 ª2010 The Authors Journal compilation ª2010 FEBS

HFE NNN110/130/234/AAA triple mutant is
glycosylation deficient
To investigate whether the three N-glycosylation sites
studied to date are the only sites of HFE N-glycosyla-
tion – and with the aim of producing an HFE mutant
that is completely deficient in N-glycosylation – we
used site-directed mutagenesis to create a NNN110/
130/234AAA triple mutant. To determine the effect of
these combined mutations on HFE, we transfected
HEK293T cells to express wild-type, single, double or
triple mutants.
Western blot analysis of cell lysates shown in Fig.3A
indicated that the triple mutant fractionated with a
lower molecular mass than the wild-type protein, and
either the single or double mutants. This lower molecu-
lar mass form appeared to be the same size as the
unglycosylated form of the wild-type protein produced
in tunicamycin-treated cultures (Fig. 3A; WT-Tunica).
These data suggested that all potential glycosylation
sites had been mutated. To confirm this, we transfected
HEK293T cells to express wild-type or triple mutant
forms of HFE and incubated the cells in the presence
and absence of tunicamycin. Drug treatment resulted
in the accumulation of an unglycosylated lower molec-
ular mass form of the wild-type HFE protein, whereas
it had no effect on the apparent molecular mass of the
triple mutant (Fig. 3B).
In a second approach, HFE was immunoprecipitated
from wild-type or triple-mutant-expressing cells and
incubated with PNGase-F. As is shown in Fig. 3C,
enzyme treatment of wild-type HFE resulted in the
production of a lower molecular mass product. Treat-
ment had no detectable effect on migration of the tri-
ple mutant, which had the same apparent molecular
mass as the PNGase F-treated wild-type protein. We
conclude that HFE is normally glycosylated in vivo at
three sites (N110, N130 and N234), and that mutation
of these sites gives rise to an HFE protein that is
N-glycosylation deficient.
N-Glycosylation of HFE is required for its
appropriate localization to the ERC
Our results from Fig. 1 indicated that tunicamycin
treatment of HFE-WT-expressing cells results in a
reticular localization pattern. To definitively establish
the importance of N-glycosylation on HFE localization
in HuTu80 cells, we transfected cells to express GFP-
tagged forms of wild-type or triple-mutant HFE. As
shown in Fig. 4A, HFE-WT–GFP localized predomi-
nantly to a tubulovesicular structure near the nucleus
with some punctate staining, similar to that observed
in Fig. 1A. The GFP-tagged triple mutant, by contrast,
predominantly displayed a reticular localization pat-
tern (Fig. 4A,B). This was similar to the expression
pattern previously observed for HFE-WT in tunica-
mycin-treated cells (Fig. 1B).
A
B
C
Fig. 2. Characterization of N-glycosylation site single and double
mutants. (A) HEK293T cells were transfected to transiently express
HFE-WT–HA, HFE-N110–HA, HFE-N130A–HA, HFE-N234A–HA, HFE-
NN110/130AA–HA, HFE-NN130/234AA–HA or HFE-NN234/110AA–
HA proteins. HFE-WT–HA expressing cells were incubated for 16 h
before lysis in the presence or absence of 1 mMtunicamycin (Tunica).
Cleared cell lysates were fractioned by 11% SDS/PAGE for immuno-
blotting with a mouse anti-HA Ig. (B) Transiently transfected HEK293T
cells expressing HFE-WT–HA, HFE-N110–HA, HFE-N130A–HA or
HFE-N234A–HA were incubated for 16 h in the presence or absence
of 1 mMtunicamycin as indicated (Tunica). HA-tagged proteins in
cleared cells lysates were detected by immunoblotting with a mouse
anti-HA Ig. (C) Transiently transfected HEK293T cells expressing HFE-
WT–HA, HFE-NN110/130AA–HA, HFE-NN130/234AA–HA or HFE-
NN234/110AA–HA protein were incubated for 16 h in the presence or
absence of 1 mMtunicamycin as indicated (Tunica). HA-tagged pro-
teins in cleared cells lysates were detected by immunoblotting with a
mouse anti-HA Ig.
L. Bhatt et al. N-Glycosylation of HFE
FEBS Journal 277 (2010) 3219–3234 ª2010 The Authors Journal compilation ª2010 FEBS 3223

























