
Structural properties of the protein SV-IV
Carlo Caporale
1
, Carla Caruso
1
, Giovanni Colonna
2,3
, Angelo Facchiano
4
, Pasquale Ferranti
4,5
,
Gianfranco Mamone
4
, Gianluca Picariello
4
, Flavia Colonna
3
, Salvatore Metafora
6
and Paola Stiuso
2,3
1
Dipartimento di Agrobiologia ed Agrochimica, Universita
´della Tuscia, Viterbo, Italy;
2
Dipartimento di Biochimica e Biofisica,
Seconda University Napoli, Italy;
3
Centro di Ricerca Interdipartimentale di Scienze Computazionali e Biotecnologiche, Napoli, Italy;
4
Istituto di Scienze dell’Alimentazione, CNR, Roma, Italy;
5
Dipartimento di Scienza degli Alimenti, Universita
`degli Studi
di Napoli ‘Federico II’, Italy;
6
Istituto Internazionale di Genetica e Biofisica, CNR, Napoli, Italy
We have investigated the molecular mechanisms that
produce different structural and functional behavior in the
monomeric and trimeric forms of seminal vesicle protein
no. 4, a protein with immunomodulatory, anti-inflamma-
tory, and procoagulant activity secreted from the rat
seminal vesicle epithelium. The monomeric and trimeric
forms were characterized in solution by CD. Details of
the self-association process and structural changes that
accompany aggregation were investigated by different
experimental approaches: trypsin proteolysis, sequence
analysis, chemical modification, and computer modeling.
The self-association process induces conformational
change mainly in the 1–70 region, which appears to be
without secondary structure in the monomer but contains
a-helix in the trimer. In vivo, proteolysis of seminal vesicle
protein no. 4 generates active peptides and this is affected
by the monomer/trimer state, which is regulated by the
concentration of the protein. The information obtained
shows how conformational changes between the mono-
meric and trimeric forms represent a crucial aspect of
activity modulation.
Keywords: monomer; proteolysis; seminal vesicle protein;
SV-IV; trimer.
SV-IV (seminal vesicle protein no. 4, according to its
electrophoretic mobility in SDS/PAGE) is a basic (pI ¼
8.9), thermostable, secretory protein of low M
r
(9758)
secreted from the rat seminal vesicle epithelium under
strict androgen transcriptional control [1–6]. SV-IV has
been purified to homogeneity and characterized exten-
sively [1–7]. We have demonstrated that this protein is a
highly flexible molecule behaving in aqueous solution as
a concentration-dependent self-associating system, with
the degree of association (monomer «dimer «trimer
equilibrium) related to its biological activity [7]. Its
polypeptide sequence is 90 amino acids long and is
encoded by a gene that has been isolated, sequenced,
and expressed in Escherichia coli [8–11]. SV-IV possesses
potent nonspecies-specific immunomodulatory, anti-
inflammatory, and procoagulant activity [12–22]. We
have demonstrated recently by electrospray MS that
10% of the native SV-IV molecules are phosphorylated
in vitro by protein kinase C and that this modification
involves only Ser58 [23]. Furthermore, we have unam-
biguously demonstrated that a Tyr36-linked phosphate
group is present in 14% of all native SV-IV molecules
[24].
SV-IV possesses a marked ability to inhibit both in vivo
and in vitro phospholipase A
2
activity and the platelet-
activating factor biosynthetic pathway [13–15]. The native
protein, transformed by transglutaminase (EC 2.3.2.13)
into a complex polymer, binds to the surface of epididymal
spermatozoa, greatly decreasing their strong immunogenic-
ity [25,26]. Although many studies have been devoted to the
functional aspects of this protein, very little is known about
its structural properties and conformational behavior in
aqueous solutions. Recent studies have shown that its
biological activities are modulated by molecular association
of the protein [7]. In this paper, we characterize the solution
structure of the monomeric and trimeric forms of SV-IV.
Experimental CD spectra were deconvoluted into secon-
dary-structural elements and compared with structural
predictions. Finally, details of the self-association process
and structural changes that accompany aggregation were
investigated by different experimental approaches: trypsin
proteolysis, sequence analysis, chemical modification, and
computer modeling.
Materials and methods
The experiments were all repeated at least four times.
Chemicals
All chemicals were of reagent grade and purchased from
BDH (Milan, Italy) or Sigma-Aldrich (Milan, Italy).
HPLC-grade solvents and reagents were obtained from
Carlo Erba (Milan, Italy). Endoproteinase Glu-C and
trypsin (sequence-grade) were from Boehringer-Mann-
heim.
Correspondence to P. Stiuso, Dipartimento di Biochimica e Biofisica,
Seconda Universita
`degli studi di Napoli, Via Costantinopoli 16,
80138-Napoli, Italy. Fax: + 39 81 5665869,
E-mail: paola.stiuso@unina2.it
Abbreviation: SV-IV, seminal vesicle protein no. 4.
(Received 20 June 2003, revised 24 September 2003,
accepted 14 November 2003)
Eur. J. Biochem. 271, 263–271 (2004) FEBS 2003 doi:10.1046/j.1432-1033.2003.03925.x

Purification of SV-IV
SV-IV was purified to homogeneity from adult rat (Wistar–
Fisher strain) seminal vesicle secretion by a previously
published technique [1]. The purity of the protein was
assessed by electrophoresis on 15% polyacrylamide gel
in denaturing and non-denaturing conditions, analysis of
amino-acid composition, fingerprint technique, and fast
atom bombardment MS [3,22]. The preparations of SV-IV
were completely free of lipopolysaccharide and tumor
necrosis factor as determined by specific biological assays
[27,28]. The concentration of the purified protein was
evaluated by its molar absorption at 276 nm
(4100
M
)1
Æcm
)1
), calculated on the basis of the tyrosine
and phenylalanine residues present in the polypeptide chain
[7].
Spectral measurements
CD measurements were performed at room temperature
with a Jasco J-720 spectropolarimeter, using quartz cells
withapathlengthof1cmand1mm.Meanresidue
ellipticities were calculated from:
½h¼MRMhobs=cd
where [h] is the mean residue ellipticity in degreesÆcm
)2
Æ
dmol
)1
,h
obs
is the observed ellipticity, MRM is the mean
residue molecular mass calculated from the sequence, dis
the optical path length (cm), and cis the concentration
in gÆmL
)1
. The CD spectra were analyzed in the region
between 200 and 250 nm to evaluate the amount of
secondary structure by using the instrument computerized
program. Spectroscopic analyses were always carried out on
dialyzed samples.
Concentration difference spectra
The difference spectra were determined by comparison of
the spectra measured with two different protein concen-
trations in two different cells. The CD spectra were
obtained at 25 C, using two cells with different light-path
lengths (L1 and L2) and filled with solutions of SV-IV in
NaCl/P
i
(0.15
M
NaCl in 0.05
M
sodium phosphate buffer,
pH 7.5). The SV-IV concentrations, C1 and C2, were
chosen in such a way that C1 ·L1 ¼C2 ·L2. In these
conditions, equal numbers of molecules are expected to be
in the light pathway at the two different concentrations
used.
Digestion of monomeric and trimeric SV-IV with trypsin
First, 25 nmol monomeric (protein concentration 0.015
mgÆmL
)1
) and trimeric (protein concentration 1.0 mgÆmL
)1
)
SV-IV were digested separately with trypsin (enzyme/
substrate, 1 : 50, w/w) at 37 C in 0.1% ammonium
bicarbonate buffer, pH 8.0. Aliquots of the incubation
mixtures, corresponding to 5 nmol of the original protein,
were then withdrawn at times ranging from 15 min to 12 h
and freeze-dried. The digests were then dissolved in 0.2 mL
aqueous 0.1% trifluoroacetic acid and resolved by RP-
HPLC on a l-Bondapak C
18
column. Eluent A was
aqueous 0.1% trifluoroacetic acid and eluent B was 0.07%
trifluoroacetic acid in acetonitrile. The elution was per-
formed at a flow rate of 1 mLÆmin
)1
using the following
program: 10 min 5% B followed by a two-step linear
gradient from 5% to 18% B over 50 min and from 18% to
28% B over 70 min. Peaks were collected manually and
freeze-dried. HPLC procedures were carried out on a
Beckman GOLD apparatus equipped with a variable-
wavelength monitor (model 166). The l-Bondapak C
18
column (0.39 ·30 cm) was from Waters-Millipore (Mil-
ford, MA, USA).
Sequence analyses
The purified tryptic peptides of monomeric and trimeric
SV-IV were dissolved in aqueous 0.1% trifluoroacetic acid
(30–60 lL); aliquots (200–500 pmol) were submitted to
sequence analysis using a pulsed liquid-phase automatic
sequencer (model 477A) equipped on-line with phenyl-
thiohydantoin amino acid analyzer (model 120A). Relevant
reagents were from Perkin Elmer/Applied Biosystems.
Samples were loaded on to a trifluoroacetic acid-treated
glass-fiber filter, coated with polybrene, and washed
according to the manufacturer’s instructions. The average
and combined repetitive amino acid yields determined by
the instrument software were not lower than 90% for each
sequenced peptide. The theoretical initial yields were not
lower than 50%.
Acetylation
Appropriate amounts of purified native SV-IV (trimeric
form, 4300 gÆmL
)1
) were acetylated in the presence of excess
acetic anhydride (6 : 1, w/v) over total amino groups, and
then purified by HPLC.
HPLC/electrospray MS
HPLC was performed using a C
18
,5lm reverse-phase
column (2.1 mm internal diameter ·250 mm; Vydac) with
a flow rate of 0.5 mLÆmin
)1
on a Kontron modular
system. The column effluent was split 1 : 25 with a Valco
tee to give a flow rate of about 20 lLÆmin
)1
into the
electrospray nebuliser. The bulk of the flow was run
through the detector for peak collection after reading of
peptide absorbance at 220 nm. Solvent A was 0.03%
trifluoroacetic acid in water (v/v); solvent B was 0.02%
trifluoroacetic acid in acetonitrile.
The electrospray device was a Platform single-quadrupole
mass spectrometer (Micromass, Manchester, UK). The
source temperature was 120 C. Mass scale calibration was
carried out using myoglobin as the reference compound.
Quantitative analysis of components was performed by
integration of the multiple charged ions of the single species.
For protein analysis, the separation was attained with a
linear gradient of 20–40% solvent B over 40 min and mass
spectra were acquired in the range 1800–500 m/z at a scan
cycle of 5 s/scan. For peptide analysis, the separation was
carried out with a linear gradient of 8–40% solvent B
over 60 min, and mass spectra were acquired in the range
1600–400 m/z at a scan cycle of 5 s/scan.
264 C. Caporale et al.(Eur. J. Biochem. 271)FEBS 2003
Endoproteinase Glu-C digestion
Endoproteinase Glu-C (Boehringer-Mannheim Italia)
hydrolytic digestion was carried out in 0.4% ammonium
bicarbonate, pH 8, at 40 C for 18 h at a substrate/enzyme
ratio of 50 : 1 (w/w). The reaction was stopped by freeze-
drying.
MALDI-TOF MS
a-Cyano-4-hydroxycinnamic acid (Fluka, Buchs, Switzer-
land) was used as matrix. The protein or peptide samples
(1 lLfromasolution1gÆL
)1
in water) were loaded on
the target and dried. Afterwards, 1 lLofa10mgÆmL
)1
solution of matrix in a mixture of 0.1% trifluoroacetic
acid in water and acetonitrile. The samples were analysed
with a Voyager DE-Pro (PerSeptive Biosystem, Framing-
ham, MA, USA) mass spectrometer operating either in
linear or in reflector mode for post source decay tandem
MS.
Structure predictions and modeling
Software and databases publicly available on the net were
used for the sequence analyses and structure predictions.
BLAST
[29] was used to search for amino-acid sequence
similarities between the SV-IV sequence and proteins
collected in databases. 3D-PSSM [30], genetTHREAD
[31], and
TOPITS
[32] were used to apply the fold recognition
strategy, searching for known protein folds compatible with
the SV-IV sequence. PHD [33], JPRED [34], and PSI-
PRED [35] web services were used to predict the secondary
structure.
The 3D model of the peptide corresponding to the
segment 70–90 of SV-IV was created by using the
INSIGHTII
package (Accelrys, San Diego, CA, USA). The Biopolymer
module was used to build the chain of amino acids, folded
as an a-helix in agreement with the secondary-structure
prediction and CD spectra results. The initial model was
geometrically optimized by energy minimization according
to the standard settings of the Optimize option.
Results
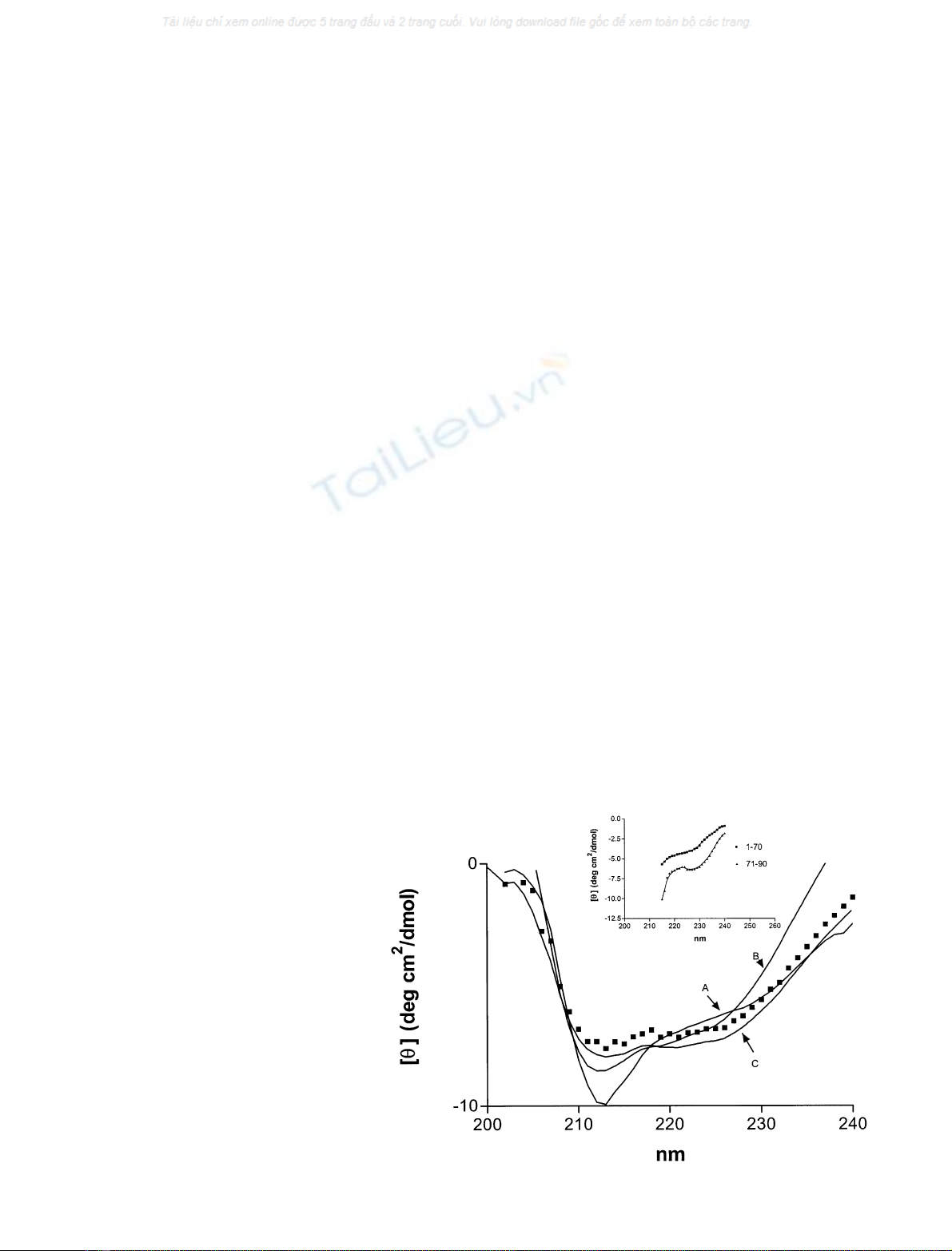
Conformational study of SV-IV
Structural modifications between the monomeric and
trimeric form of the SV-IV protein are evident on CD
spectra (Fig. 1). The far-UV CD spectra, as characterized
by an isodichroic point located at about 208 nm indicate the
presence of two-state equilibria between the monomeric and
trimeric forms. The self-association process was accompan-
ied by structural changes in the protein. The secondary-
structure analysis program estimated that the a-helix
content is 24% in the monomeric form and 45% in the
trimeric form, and b-structure is absent from both forms.
The CD spectra of the 1–70 and 70–90 regions of the protein
are reported in the inset of Fig. 1. The secondary-structure
analysis program estimated about 100% a-helix for the
70–90 fragment and 2–3% for the 1–70 segment. This
suggests that the C-terminal segment of the protein is
organized as a-helix in the whole protein, and the 1–70
region is poorly structured.
SDS increases the a-helix content of proteins revealing
the helical potential. The a-helix content of the monomeric
form of SV-IV is approximately doubled by the addition of
5.4 m
M
SDS (Fig. 1), whereas in the 1–70 region, the a-helix
content increases from 2–3% to 23% in the presence of
SDS (data not shown). This suggests that the effect of SDS
on the whole protein is exerted in the 1–70 region, which
probably plays a fundamental role in the self-association
process, with secondary-structure reorganization occurring
in going from the monomeric to the trimeric form.
Predictive methods have been applied to obtain a
theoretical model of the structural organization of SV-IV
protein. The amino-acid sequence was analyzed using the
BLAST
program to find similar proteins in the nrdatabase
(nonredundant database consisting of all protein sequences
Fig. 1. Structural characterization of the
monomeric (0.01 lgÆlL
)1
) and trimeric
(0.1 lgÆlL
)1
) form of SV-IV protein. Far-UV
CDspectraatdifferentconcentrationsofthe
protein (A, 0.01 lgÆlL
)1
monomeric form;
B, 0.05 lgÆlL
)1
monomeric/trimeric mixture;
C, 0.1 lgÆlL
)1
trimeric form). Monomeric
form in SDS (j,5.4m
M
). The CD spectra of
the 1–70 and 70–90 fragment of SV-IV are
reported in the inset. Each spectrum represents
an average of five scans. The SV-IV samples
are in 50 m
M
Tris/HCl, pH 7.2.
FEBS 2003 Structural properties of SV-IV (Eur. J. Biochem. 271) 265
present in the databases). No protein of known 3D structure
was found to have sequence similarity suitable to apply the
homology modeling strategy, i.e. at least 20–30% sequence
identity. As an alternative, the fold-recognition approach
was applied by using three independent servers on the net:
3D-PSSM, GenetTHREAD, TOPITS. None of the meth-
ods identified a known fold suitable for modeling the SV-IV
protein or the 1–70 region. Therefore, in conclusion, the two
most reliable strategies for predicting the 3D model of a
protein, i.e. homology modeling and fold-recognition strat-
egies, were unable to create a model for either the whole
SV-IV protein or the 1–70 region, and this suggests that this
protein assumes a global structure that is not similar to any
protein of known 3D structure.
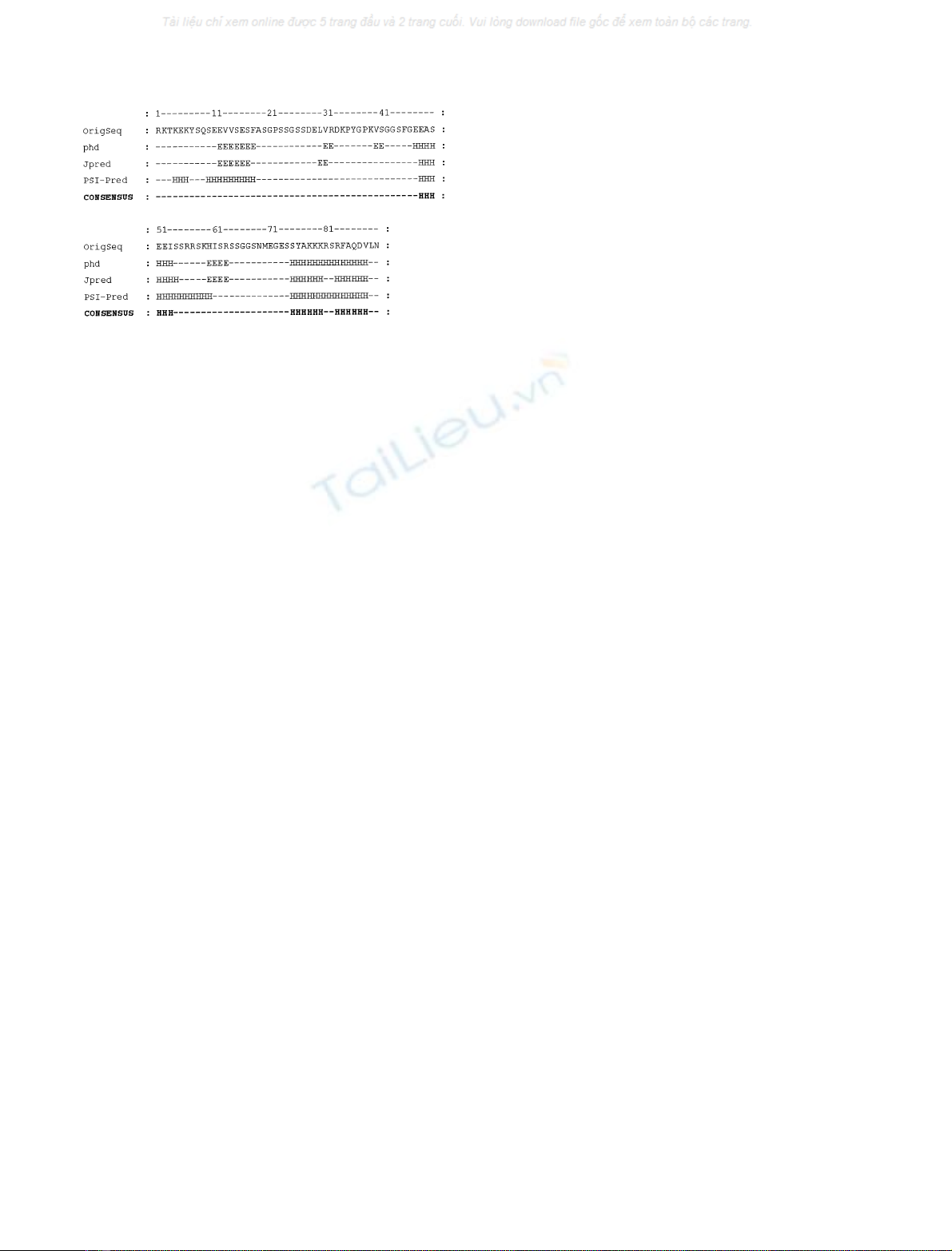
Secondary-structure predictions were performed by dif-
ferent methods, i.e. JPRED, PHD, PSI-PRED (Fig. 2). A
consensus predictionbased on the agreement between
different methods can be considered more successful than
the single method used. The consensus prediction suggests a
few helical regions (48–53 and the C-terminal region)
covering 20% of the protein, which is in good agreement
with the secondary-structure content revealed by CD studies
of 24% a-helix for the monomeric form.
Protein digestion and fragment characterization
SV-IV is a 90-amino-acid protein lacking disulfide bridges
and possessing nine lysine and seven arginine residues,
which represents a large number of potential hydrolysis sites
for trypsin. For this reason, we selected this protease to
investigate the different accessibility of crucial sites sup-
porting molecule aggregation and characterizing the mono-
meric and trimeric forms. Both forms of SV-IV were
digested separately using the same enzyme/substrate ratio.
Aliquots of the incubation mixtures were withdrawn at
various times, and the formation of fragments was moni-
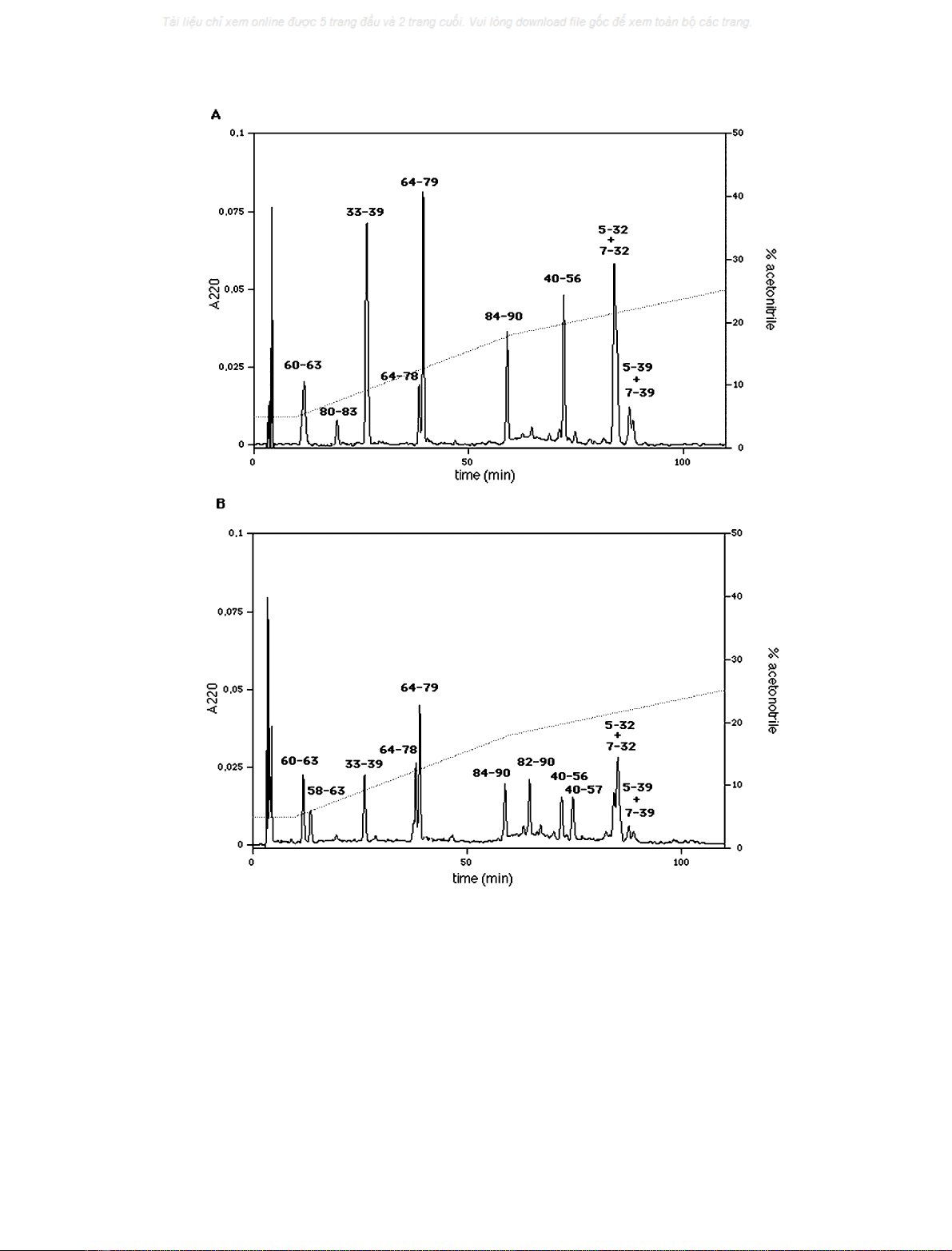
tored by RP-HPLC. Figure 3 shows chromatograms of the
digestion mixtures of the trimeric (Fig. 3A) and monomeric
(Fig. 3B) forms after 12 h incubation. Lower amounts of all
the peptides were also produced after 15 min incubation,
indicating that both monomeric and trimeric forms were
readily digested by trypsin (not shown). Each fragment
collected was submitted to automatic sequence analysis. The
corresponding start-end position in the protein sequence is
indicated in the figure. Some differences in the hydrolytic
pathways were found. The protein is hydrolyzed at Arg57
only in the monomeric form. In fact, whereas fragments
40–56 and 60–63 are common to both chromatograms,
fragments 40–57 and 58–63 arose only from the monomeric
form (Fig. 3B). The tripeptide 57–59 complementing frag-
ments 40–56 and 60–63 which originated from digestion
of the trimer was not identified in the chromatogram
(Fig. 3A). Furthermore, fragment 80–83, complementary to
fragments 64–79 and 84–90, was generated only from
hydrolysis of the trimeric form (Fig. 3A), whereas fragment
82–90 arose only from the monomeric form (Fig. 3B),
indicating further digestion at the level of Arg81. The
dipeptide Lys80–Arg81, complementary to fragments 64–79
and 82–90, was not identified in the chromatogram
(Fig. 3B). Both peptide bonds Lys80–Arg81 and Arg81–
Ser82 seem to be hidden in the trimer, as the whole fragment
Lys80-Arg81-Ser82-Arg83 was found (Fig. 3A), whereas no
fragment ending at Lys80 or starting at Arg81 arose from
digestion of the monomer (Fig. 3B). As a consequence,
Arg81 should be accessible only in the monomeric form,
whereas Lys80 also seems to be quite hidden in the
monomeric form.
SV-IV is not fully acetylated by acetic anhydride
An aliquot was directly analysed by HPLC/electrospray MS
to characterize the acetylated form of SV-IV protein. As
shown by its transformed spectrum (Fig. 4), several com-
ponents were present, differing with respect to the number
of acetyl groups (mass increase of 42 for each acetyl group
incorporated), and indicating that the reaction was not
complete, but generated a mixture of incompletely acetyl-
ated forms of the protein. These contained from three to
eight acetyl groups (the maximum expected was 10,
considering nine lysine residues and the N-terminal amino
group), the most abundant ranging from four to six.
To identify the acetylated residues, another aliquot of
protein was first digested with endoproteinase Glu-C and
then analysed by HPLC/electrospray MS to obtain the
relevant peptide map. The peptides identified are shown in
Table 1. We were therefore able to screen the whole protein
sequence to identify the acetylated peptides.
Peptides were identified by their molecular mass on the
basis of the known protein sequence and the endoproteinase
Glu-C specificity. In most cases, a mixture of the native and
acetylated peptides was observed and identified by the mass
increase of 42 mass units. The relative level of acetylation of
a peptide was estimated on the basis of the intensity ratio of
the native and acetylated species. From the data summar-
ized in Table 1, it can be seen that some of the peptides were
almost completely acetylated whereas some showed low or
minimal acetylation. To locate acetylated Lys residues on
peptides containing more than one Lys, the fractions
collected from the HPLC separation (Fig. 6) were analysed
by tandem MS using MALDI-TOF PSD-MS. As an
example, peptide 6–12, containing Lys6, was acetylated only
to 5%; peptide 72–90, containing Lys78, Lys79, and Lys80,
showed partial acetylation at one of the three residues,
because the signal of the triacetylated species was less
intense than that of the diacetylated species. The MS/MS
Fig. 2. Secondary-structure prediction. Amino-acid sequence and sec-
ondary-structure predictions performed with PHD, JPRED, and PSI-
PRED (see Materials and methods). a-Helix is indicated by H and
b-strand conformation is indicated by E.
266 C. Caporale et al.(Eur. J. Biochem. 271)FEBS 2003
analysis of the chromatographic fraction showed that the
two Lys residues at positions 78 and 79 were both
acetylated, whereas Lys80 was not (Fig. 5).
Molecular modeling of 70–90 region
CD spectra of SV-IV fragment 70–90 and the secondary-
structure prediction suggested that the region 70–90 should
have a high a-helix content. We created a computer model
of the 70–90 peptide. The initial conformation of the
backbone was imposed as a-helix, and energy minimization
was performed in order to optimize the peptide structure. As
a consequence of such optimization, the initial backbone
conformation was approximately conserved only in the
region corresponding to the 70–81 segment (Fig. 6). In the
82–90 segment, a helical conformation was conserved, but it
was not consistent with a-helix features, as the Kabsch and
Sander assignment of secondary structure did not define
helix in this segment of the peptide. The initial conformation
of side chains was also modified under energy minimization,
Fig. 3. Protein digestion. Chromatograms of digestion mixtures of trimeric (A) and monomeric (B) forms of SV-IV after 12 h incubation with
trypsin. Each peak is labelled with the corresponding protein fragment. Peaks present in both chromatograms refer to the protein segments 60–63,
33–39, 64–78, 64–79, 84–90, 40–56, 5–32 + 7–32, 5–39 + 7–39. Peaks present in chromatogram A but not in B refer to segment 80–83. Peaks
present in chromatogram B but not in A refer to the protein segments 50–63, 82–90, 40–57.
FEBS 2003 Structural properties of SV-IV (Eur. J. Biochem. 271) 267

























