
MINIREVIEW
Substrate recognition by the protein disulfide isomerases
Feras Hatahet and Lloyd W. Ruddock
Biocenter Oulu and Department of Biochemistry, University of Oulu, Finland
Introduction
The compartmentalization of cells is essential for life.
Each subcellular compartment has highly defined func-
tions and is highly regulated in terms of biophysical
conditions (pH, ion concentration, etc.), small mole-
cule composition, and protein composition. The endo-
plasmic reticulum (ER) is a compartment with
multiple functions, including steroid production [1,2],
carbohydrate metabolism, including glycogenolysis [3],
transformation of bile pigments [4,5] detoxification of
many drugs [6], calcium storage [7,8] and phospholipid
synthesis [9,10]. However, the primary function of the
ER is usually deemed to be the folding and post-trans-
lational modification of proteins destined for secretion,
the outer membrane and compartments in the secre-
tory pathway such as the Golgi.
Over one-third of all human proteins fold in the ER
[11], and it contains many catalysts that generate
unique post-translational modifications. These range
from the well known, such as disulfide bond formation
[12] and N-glycosylation [13], to the less well known,
such as the formation of formyl-glycine in the active
site of the sulfatases [14,15] or c-carboxyglutamate for-
mation in blood clotting factors [16].
Disulfide bonds are covalent linkages formed
between two cysteine residues in proteins whose pri-
mary function is to stabilize the folded structure of the
protein. As any cysteine residue in a protein has the
potential to form a disulfide bond with another cyste-
ine, either intramolecular or intermolecular, the correct
formation of native disulfide bonds is often the rate-
limiting step in the folding of proteins in vitro and
in vivo [17,18]. Disulfide bond formation is not unique
to the ER; it also occurs in the periplasm of Gram-
negative bacteria [19], in mitochondria [20,21], and
outside the cell, e.g. in blood clotting [22], and there
are a small, but growing, number of examples of
Keywords
diagonal electrophoresis; disulfide bond;
endoplasmic reticulum; glutathione;
molecular chaperone; oxidative protein
folding; protein folding catalyst; substrate
binding
Correspondence
L. W. Ruddock, Biocenter Oulu and
Department of Biochemistry, PO Box 3000,
90014 University of Oulu, Finland
Fax: +358 8 553 1141
Tel: +358 8 553 1683
E-mail: Lloyd.ruddock@oulu.fi
(Received 8 June 2007, revised 27 July
2007, accepted 17 August 2007)
doi:10.1111/j.1742-4658.2007.06058.x
Protein folding in the endoplasmic reticulum is often associated with the
formation of native disulfide bonds. Their primary function is to stabilize
the folded structure of the protein, although disulfide bond formation can
also play a regulatory role. Native disulfide bond formation is not trivial,
so it is often the rate-limiting step of protein folding both in vivo and
in vitro. Complex coordinated systems of molecular chaperones and protein
folding catalysts have evolved to help proteins attain their correct folded
conformation. This includes a family of enzymes involved in catalyzing
thiol–disulfide exchange in the endoplasmic reticulum, the protein disulfide
isomerase (PDI) family. There are now 17 reported PDI family members in
the endoplasmic reticulum of human cells, but the functional differentiation
of these is far from complete. Despite PDI being the first catalyst of
protein folding reported, there is much that is still not known about its
mechanisms of action. This review will focus on the interactions of the
human PDI family members with substrates, including recent research on
identifying and characterizing their substrate-binding sites and on determin-
ing their natural substrates in vivo.
Abbreviations
ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; GSSG, oxidized glutathione; PDI, protein disulfide
isomerase.
FEBS Journal 274 (2007) 5223–5234 ª2007 The Authors Journal compilation ª2007 FEBS 5223

regulatory disulfide bond formation in the cytoplasm
[23] or of, perhaps, extensive disulfide bond formation
in the cytoplasm of thermophiles [24]. Although disul-
fide bond formation is not unique to the ER, the com-
plexity of the system, in terms of the number of
substrate proteins that need the addition of disulfide
bonds and the number of enzymes involved in catalyz-
ing the process, is unique.
Disulfide bond formation in the ER
The process of native disulfide bond formation in the
ER is known to be catalyzed by several families of
enzymes. However, although some of the participants
in the cellular process are known, their precise roles
and mechanisms of action are still largely unknown.
Native disulfide bond formation can occur via multiple
parallel pathways, and this significantly complicates
the interpretation of in vivo data. For example, there
appear to be at least five potential parallel pathways
for oxidation (disulfide bond formation) in substrate
proteins. The first is direct oxidation of dithiols in
folding proteins by molecular oxygen )however, this
reaction, unless catalyzed, is probably too slow to be
physiologically relevant. The second is oxidation by low
molecular weight biochemical compounds such as oxi-
dized glutathione (GSSG) [25–27]. The third is oxida-
tion catalyzed by proteins belonging to the thioredoxin
superfamily, such as protein disulfide isomerase (PDI)
[28,29]. However, as highlighted in an excellent recent
review [30], strictly speaking, neither the PDIs nor
GSSG by themselves have oxidase activity, as neither
utilizes molecular oxygen and both are converted into
the reduced form by their donation of a disulfide bond
to a substrate protein; hence, both need to be
reoxidized to complete the catalytic cycle. The fourth
is oxidation by flavin-dependent sulfhydryl oxidases
[31,32]. However, there is little supporting evidence for
a normal physiological role of the QSOX sulfhydryl
oxidases in disulfide bond formation in the ER [33],
whereas the active site of yFMO [31] probably faces
the cytosol and not the ER lumen. The fifth is oxida-
tion by Ero1, although the evidence to date [34–37]
suggests that this occurs indirectly via PDI family
members, with no evidence of direct oxidation of
substrate proteins. Although the gene products of
ERO1 and PDI1 are essential for viability in yeast
[38,39] and those of the other components, including
the glutathione biosynthetic pathway, are not, it is still
unclear to what extent the five possible oxidative path-
ways contribute to native disulfide bond formation
under physiologically normal conditions. What is clear
is that the rate-limiting step for native disulfide bond
formation in proteins that contain multiple disulfides
comprises late-stage isomerization reactions, where
disulfide bond formation is linked to conformational
changes in protein substrates with substantial regular
secondary structure. These steps are only catalyzed
by members of the PDI family, and hence knowl-
edge of the mechanisms of action of these proteins is
critical for our understanding of native disulfide bond
formation.
PDI was the first protein-folding catalyst reported
(over 40 years ago [40]), but as yet significant details
of its mechanism of action are unknown. PDI is a
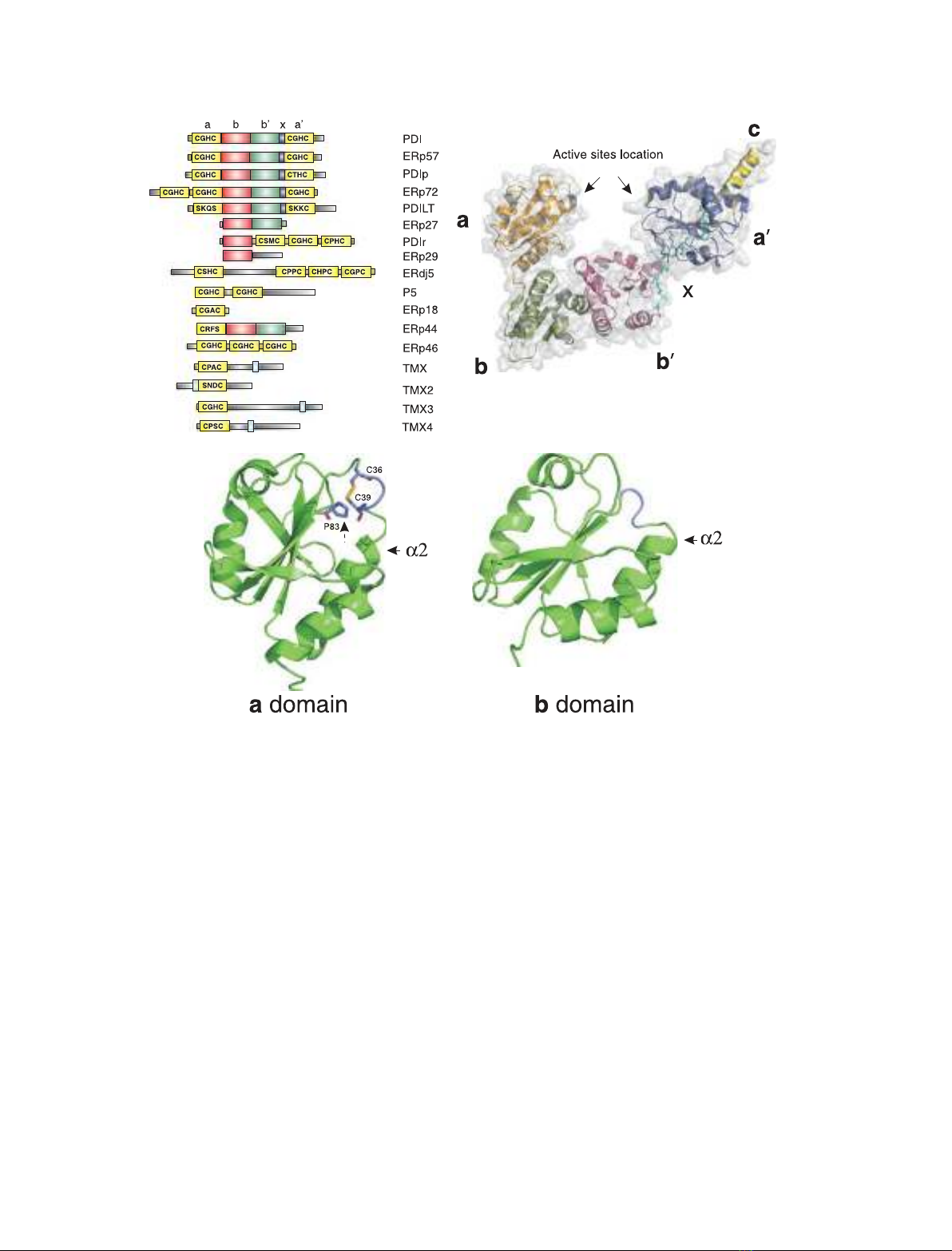
multidomain protein with two catalytic domains, aand
a¢, which are separated by two noncatalytic domains b
and b¢. In addition, there is a 19 amino acid linker
region between b¢and a¢, designated x, and a highly
acidic C-terminal extension, designated c, which con-
tains the C-terminal ER-localization motif KDEL and
that has a low affinity for Ca
2+
, but that seems to
have no function in the catalysis of native disulfide
bond formation by PDI (Fig. 1). The structures of the
a-domain, b-domain and a¢-domain of human PDI
have been solved by NMR [41,42] and (PDB file
1X5C, N Tochoi, S Koshiba, M Inoue, T Kigwa & S
Yokoyama, unpublished results), but both the isolated
b¢-domain of human PDI and all multidomain cata-
lytic PDI family members have proved intransigent to
structure resolution for over 30 years, with the recent
exception of yeast Pdi1p [43] (Fig. 1B). Over the past
decade, the number of PDI family members in mam-
malian cells has grown, and there are now 17 reported
family members in humans [28], two of which, ERp57
and PDIp, share the same domain architecture as PDI
(Fig. 1). With the exception of the first domain of
ERp29 [44], all of the PDI family member domains
solved to date share the thioredoxin fold, an ab-fold
with a mixed b-sheet core [41–43,45,46] and (PDB files
1X5C, 1X5D, 1X5E, 2DIZ, 2DJ1, 2DJ2, 2DJ3, 2DML
and 2DMM, N Tochoi, S Koshiba, M Inoue, T Kigwa
& S Yokoyama; ISEN, ZJ Liu, L Chen, W Tempel, A
Shah, D Lee, JP Rose, DC Richardson, JS Richardson
& BC Wang, unpublished results). In the catalytically
active domains, the CXXC active site, which exists in
dithiol, disulfide and mixed disulfide states during the
catalytic cycle, lies at the N-terminus of a2. Spatially
juxtaposed is a cis-proline (Fig. 1C) that lies before b4
and that is implicated in substrate interactions in other
thioredoxin superfamily members [47,48]. Despite the
name, five of the 17 human PDI family members,
PDILT, ERp27, ERp29, ERp44, and TMX2, lack a
CXXC active site and therefore may not be involved in
catalyzing native disulfide bond formation. This poten-
tial confusion over the family name arises because the
PDI–substrate interactions F. Hatahet and L. W. Ruddock
5224 FEBS Journal 274 (2007) 5223–5234 ª2007 The Authors Journal compilation ª2007 FEBS
PDI family is a subfamily of the thioredoxin superfam-
ily, which is defined by subcellular localization, i.e. by
being in the ER, rather than by function.
In addition to having a role in protein folding in the
ER, PDI is the b-subunit of prolyl-4-hydroxylase [49]
and microsomal triglyceride transfer protein [50], and
A
C
B
Fig. 1. The architecture of the PDI family. (A) Domain structures of the human PDI family. PDI has a four-domain structure, the catalytic
a-domain and a¢-domain being separated by the noncatalytic b-domain and b¢-domain. Whereas the b¢-domain provides the primary sub-
strate-binding site of human PDI, no physiological function has been assigned to the b-domain, and it is probably structural in nature.
Whereas the linkers between the a-domain and b-domain and between the b-domain and b¢-domain are very short, the b¢-domain and
a¢-domain are linked by a 17 amino acid linker designated x[81]. Other PDI family members either have a similar overall domain architecture
to that of PDI, e.g. ERp57 and PDIp, or have different combinations of catalytic and noncatalytic domains. (B) X-ray structure of Saccharomy-
ces cerevisiae Pdi1p. The only full-length structure of a catalytically active PDI family member reported is that of S. cerevisiae Pdi1p [44].
Each of the four domains of Pdi1p, a,b,b¢and a¢, shows a thioredoxin fold. The active site motifs (marked with arrows) in the a-domain
and a¢-domain lie close to each other. In the reported structure, the b-domain of one molecule of Pdi1p is located in the putative substrate-
binding pocket formed between the other four domains of another molecule of Pdi1p; this may have resulted in increased rigidity and have
allowed the crystal to form. (C) NMR structures of the a-domain and b-domain of human PDI. The a-domain of human PDI [41] shares the
same fold as the archetypal member of the superfamily, thioredoxin. It has a five-stranded mixed b-sheet surrounded by four a-helices. The
active site motif (C36 and C39) lies at the N-terminus of a2, and the helix dipole is thought to contribute to the nucleophilicity of the N-termi-
nal active site cysteine. There is also a conserved cis-proline in the structure (marked with a dashed arrow) located before b4. This residue
has been implicated in substrate binding within the superfamily [50,51]. Despite having virtually no sequence similarity with the a-domain,
the b-domain [42] also has a thioredoxin fold, although it lacks the active site and the cis-proline. The substrate-binding site in the b¢-domain
(which is homologous to the b-domain) is located at the same position as the active site in the a-domain and a¢-domain. The structure of the
a¢-domain (PDB file 1X5C, N Tochoi, S Koshiba, M Inoue, T Kigwa & S Yokoyama, unpublished results) is not shown, but it is similar to that
of the a-domain.
F. Hatahet and L. W. Ruddock PDI–substrate interactions
FEBS Journal 274 (2007) 5223–5234 ª2007 The Authors Journal compilation ª2007 FEBS 5225

PDI family members are involved in ER-associated
degradation (ERAD [51–53]), although it is still
unclear which family member(s) plays the primary role
in disulfide reduction in proteins targeted for degrada-
tion, and in retrotranslocation of bacterial toxins
[54,55] and viruses [56], which hijack normal cellular
processes to exert their biological effect. In addition,
PDI family members have been reported to be
involved in a wide range of biological processes
located in nearly every cellular compartment [57]. The
role of PDI family members in these processes, which
include sperm–egg fusion, blood clotting, and integrin
ligation [58–60], is often determined by the use of
inhibitors, which may not be specific for only one fam-
ily member, and by the use of inhibitory antibodies,
which have often not been tested for crossreactivity.
Hence, in many cases it is not completely clear which
PDI family member is involved. In all cases, further
clarification is required as to how the PDI family
member escapes the ER, as most have consensus C-ter-
minal ER-localization motifs; for example, PDI,
ERdj5, P5 and ERp46 have KDEL. Furthermore, the
interaction of substrates with PDI family members is
one of the mechanisms involved in keeping non-native
proteins in the ER [61,62]. This question of nonexpect-
ed localization is especially relevant for nonsecretory
pathway compartments, as there are no known experi-
mentally confirmed splice variants of PDI family mem-
bers that lack the N-terminal ER signal sequence.
To understand the mechanisms of action of the PDI
family, we must understand the nature of the interac-
tion(s) between PDI family members and their sub-
strates, including the specificities of substrate binding.
However, substrate binding is complex, in part because
PDI family members appear to have very broad speci-
ficities and in part because they have multiple sub-
strate-binding sites. It should be noted that although
the specificity of substrate binding and the specificity
of the catalysis of thiol–disulfide exchange are inter-
linked, they are not the same thing. Specifically, the
rate of catalysis of thiol–disulfide exchange by PDI
family members is not directly linked to substrate-
binding affinity, and each PDI family member may be
able to bind substrates in which it is unable to catalyze
thiol–disulfide exchange. In addition to binding protein
substrates, at least one PDI family member must be
able to trigger conformational changes in bound non-
native protein subtrates to allow access to buried
disulfides or free thiols in folding intermediates with
substantial regular secondary structure. This is an area
that has been poorly studied, due to the extreme
difficulty in isolating ⁄identifying intermediates and
following the process.
Folding catalyst or molecular
chaperone?
Since the experiments by Anfinsen [63], it is generally
accepted that the final structure of a protein is deter-
mined solely by the primary sequence of the protein,
plus post-translational modifications, and the physical
properties of the solution, such as pH and ionic
strength. However, for the majority of proteins, there
are auxiliary molecules inside the cell that help the
protein to attain this final conformation. These fall
into two broad types: protein-folding catalysts and
molecular chaperones. Protein-folding catalysts acceler-
ate slow steps in the productive folding pathway. In
contrast, the net effect of molecular chaperones is to
inhibit steps in nonproductive folding pathways. For
example, they prevent partially folded intermediates
from aggregating, or delay the initiation of folding
until appropriate sequences have been translated. PDI
is a protein-folding catalyst that accelerates the forma-
tion of native disulfide bonds, whose noncatalyzed for-
mation, even in a physiological redox buffer, can take
days. PDI is also reported to have molecular chaper-
one activity [64,65], as it is able to prevent the aggrega-
tion of proteins that do not contain cysteine residues.
Often, these activities are seen as being distinct, but
they should not be. For PDI family members to work
as protein-folding catalysts, they require molecular
chaperone-like properties, i.e. the ability to recognize
and interact with non-native proteins and so allow
catalysis of disulfide bond formation, including gaining
access to buried thiols and disulfide bonds, and simul-
taneously preventing non-specific interactions between
partially folded intermediates.
Molecular chaperones and protein-folding catalysts
have the ability to bind a very large number of differ-
ent substrates. For example, approximately 7500 pro-
teins fold in the human ER [11], the majority of which
require native disulfide bond formation and hence
probably interact with a catalytically active PDI family
member. There is an added complexity in that for each
folding protein there will be multiple folding intermedi-
ates, from ‘unfolded’ through to quasi-native states.
For the well-studied folding pathway of bovine pancre-
atic trypsin inhibitor [17,66,67], PDI is able to catalyze
all of the steps in the folding reaction, implying that it
recognizes all folding intermediates in the pathway.
Whereas the ER lectin-based molecular chaperones,
such as calnexin and calreticulin, have a highly specific
recognition motif, binding only monoglucosylated
N-glycans [68,69], those chaperones that bind protein
substrates directly appear to have much broader speci-
ficities. For example, using a peptide phage display
PDI–substrate interactions F. Hatahet and L. W. Ruddock
5226 FEBS Journal 274 (2007) 5223–5234 ª2007 The Authors Journal compilation ª2007 FEBS

library, Gething and coworkers were able to demon-
strate that BiP, the ER-resident HSP70, recognized
sequences with the motif HyXHyXHyXHy (where Hy
is a large hydrophobic amino acid) and in particular
the sequence (WF)X(WF)X(WFLI)X(WFYL) [70]; this
sequence can be found in 1032 human proteins. One
common feature of molecular chaperones is that they
bind substrates with relatively low affinity. For exam-
ple, BiP binds synthetic peptides with consensus motifs
with 10–60 lmaffinity [70]. A recent landmark paper
monitoring chaperone engagement of substrates in the
ER of live cells [71] elegantly showed that chaperone
substrate interactions in vivo are very dynamic, with
rapid sampling and release. In this context, it is also
important to remember that equivalent affinities do
not mean equivalent dynamics. For example, the affin-
ity of calreticulin for substrate is about 2 lm, and that
of calreticulin for ERp57 is comparable at around
18 lm, but the exchange rate of K
off
for N-glycopro-
teins is 10
5
-fold slower than that for ERp57 [72,73].
The affinity of PDI for substrates is comparable to
that of molecular chaperones [74,75], with the highest
affinity for a noncysteine-containing peptide substrate
known to the authors being about 0.25 lm(A Pirnes-
koski and LW Ruddock, unpublished results). This
low affinity and the high dynamics make it difficult to
study PDI–substrate interactions in complex mixtures
or in vivo unless the proteins are physically joined
together by the addition of chemical crosslinkers. The
use of chemical crosslinkers, although potentially
prone to artefacts, has allowed the identification of
multiple binding sites on human PDI family members
and determination of the specificity of one family
member (see below).
The substrate-binding sites of PDI
Initial attempts to identify the substrate-binding site
in PDI family members were based on the interaction
of domain constructs of human PDI expressed rec-
ombinantly in Escherichia coli with small peptide
ligands such as D-somatostatin and with non-native
proteins such as scrambled RNase. The results indi-
cated that the noncatalytic b¢-domain was essential
and sufficient for the binding of small peptides,
whereas the catalytic a-domain and a¢-domain also
contributed to the binding of larger peptides and
non-native proteins [76]. These interactions were pri-
marily hydrophobic in nature, consistent with a role
in binding non-native proteins. These results correlate
well with studies published around the same time on
the activities of equivalent domain constructs, which
showed that thiol–disulfide exchange could be cata-
lyzed by any construct containing either the
a-domain or a¢-domain, that disulfide isomerization
required a linear combination of one of these cata-
lytic domains plus the b¢-domain, and that complex
isomerization, i.e. isomerization in protein substrates
with substantial regular secondary structure, required
the whole protein excluding the c-region [77]. Hence,
the b¢-domain is required to bind non-native protein
substrates with reasonable affinity to allow the cata-
lytic domains to act on them. Follow-up studies on
the b¢-domain binding site by modeling and mutagen-
esis identified the binding site as being in a homolo-
gous location to the catalytic site in the a-domain
and a¢-domain (Fig. 1B) [78]). Furthermore, parallel
studies determined: (a) that the binding specificity of
the b¢-domain in PDIp was a single tyrosine or tryp-
tophan with no adjacent negative charge [79];
(b) that the specificity of PDIp for substrates could
be modulated by mutating analogous residues to
those that formed the substrate-binding site of PDI
(KEH Salo and LW Ruddock, unpublished results);
(c) that this binding site in the b¢-domain of ERp57
had become specialized for the interaction of ERp57
with the P-domains of calreticulin and calnexin [80];
and (d) that an overlapping site in the second
domain of ERp29 and the Drosophila ERp29 homo-
log was used to bind substrates [81,82].
Since the first publication on the substrate-binding
properties of the b¢-domain, this site has been referred
to as the primary substrate-binding site of PDI, imply-
ing that it is not the only site of interaction between
PDI family members and their substrates. There are
three pieces of evidence indicating that there are
secondary substrate-binding sites in the a-domain and
a¢-domain. First, the isolated a-domain and a¢-domain
are able to catalyze thiol–disulfide exchange in small
peptide substrates, for which they must have a reason-
able affinity [83]. Secondly, in vivo crosslinking studies
indicated that the binding of larger peptides required a
linear combination that included both b¢and aor a¢,
and the efficient crosslinking of non-native proteins
required the whole protein except the c-region [76].
Third, studies on the interaction of PDI with the
a-subunit of prolyl-4-hydroxylase to form a stable
active tetramer allowed the identification of interaction
sites in the a-domain, b¢-domain and a¢-domain of
PDI, with the site in the b¢-domain being equivalent to
the primary substrate-binding site [84]. Parallel studies
with the binding of non-native proteins implicated the
same sites in the a-domain, b¢-domain and a¢-domain
in PDI substrate binding (HI Alanen and LW Rud-
dock, unpublished results). The site identified in the
a¢-domain partially overlaps with a tripeptide-binding
F. Hatahet and L. W. Ruddock PDI–substrate interactions
FEBS Journal 274 (2007) 5223–5234 ª2007 The Authors Journal compilation ª2007 FEBS 5227

























