
Structural studies of thymidine kinases from
Bacillus anthracis and Bacillus cereus provide insights
into quaternary structure and conformational changes
upon substrate binding
Urszula Kosinska
1
, Cecilia Carnrot
2
, Michael P. B. Sandrini
3
, Anders R. Clausen
3
, Liya Wang
2
,
Jure Piskur
3
, Staffan Eriksson
2
and Hans Eklund
1
1 Department of Molecular Biology, Swedish University of Agricultural Sciences, Uppsala Biomedical Centre, Sweden
2 Molecular Biosciences, Swedish University of Agricultural Sciences, Uppsala Biomedical Centre, Sweden
3 Department of Cell and Organism Biology, Lund University, Sweden
Bacillus anthracis and Bacillus cereus are two closely
related species of the genus Bacillus. B. anthracis cau-
ses anthrax, a disease that in most cases has fatal
consequences [1]. B. cereus is a human pathogen asso-
ciated with food poisoning [2]. Both species produce
endospores under stressful conditions as a means
of survival through environmental stress. The major
genetic difference between these two species is associ-
ated with two toxin-encoding plasmids, pXO1 and
pXO2 [3,4], which are present in B. anthracis but not
in B. cereus.
Thymidine kinase (TK; EC 2.7.1.21) is a deoxyribo-
nucleoside kinase (dNK) that phosphorylates thymi-
dine to thymidine monophosphate. Mammals possess
Keywords
deoxythymidine triphosphate; dimer;
feedback inhibitor; phosphate donor;
tetramer
Correspondence
H. Eklund, Swedish University of
Agriculturla Sciences, Box 590, BMC,
Uppsala SE-75124, Sweden
E-mail: hasse@xray.bmc.uu.se
(Received 28 August 2006, revised 17
November 2006, accepted 24 November
2006)
doi:10.1111/j.1742-4658.2006.05617.x
Thymidine kinase (TK) is the key enzyme in salvaging thymidine to pro-
duce thymidine monophosphate. Owing to its ability to phosphorylate
nucleoside analogue prodrugs, TK has gained attention as a rate-limiting
drug activator. We describe the structures of two bacterial TKs, one from
the pathogen Bacillus anthracis in complex with the substrate dT, and the
second from the food-poison-associated Bacillus cereus in complex with the
feedback inhibitor dTTP. Interestingly, in contrast with previous structures
of TK in complex with dTTP, in this study dTTP occupies the phosphate
donor site and not the phosphate acceptor site. This results in several con-
formational changes compared with TK structures described previously.
One of the differences is the way tetramers are formed. Unlike B. anthracis
TK, B. cereus TK shows a loose tetramer. Moreover, the lasso-domain is
in open conformation in B. cereus TK without any substrate in the active
site, whereas in B. anthracis TK the loop conformation is closed and
thymidine occupies the active site. Another conformational difference lies
within a region of 20 residues that we refer to as phosphate-binding b-hair-
pin. The phosphate-binding b-hairpin seems to be a flexible region of the
enzyme which becomes ordered upon formation of hydrogen bonds to the
a-phosphate of the phosphate donor, dTTP. In addition to descriptions of
the different conformations that TK may adopt during the course of reac-
tion, the oligomeric state of the enzyme is investigated.
Abbreviations
Ba-TK, Bacillus anthracis thymidine kinase; Bc-TK, Bacillus cereus thymidine kinase; Ca-TK, Clostridium acetobutylicum thymidine kinase;
dCK, deoxycytidine kinase; dGK, deoxyguanosine kinase; dNK, deoxyribonucleoside kinase; hTK1, human thymidine kinase 1; MPD,
2-methyl-2,4-pentadiol; P-b-hairpin, phosphate-binding b-hairpin; TK, thymidine kinase; Uu-TK, Ureaplasma urealyticum thymidine kinase.
FEBS Journal 274 (2007) 727–737 ª2006 The Authors Journal compilation ª2006 FEBS 727

four deoxyribonucleoside-specific dNKs: cytosolic
deoxycytidine kinase (dCK) and TK1 and mitochond-
rial deoxyguanosine kinase (dGK) and TK2. Bacteria,
however, have a smaller group of dNKs. Most
bacteria have TK that both sequence wise and struc-
turally resembles TK1 [5]. In addition, one or two
non-TK1-like dNKs can be found in most Gram-
positive bacteria. Besides TK, there are two
dCK ⁄dGK-like dNKs in B. anthracis and B. cereus.
The amino-acid sequence identity between the TKs
from B. anthracis (Ba-TK) and B. cereus (Bc-TK) is
as high as 96%. The sequence identity with human
TK1 (hTK1) is 37–38%.
From amino-acid sequence analysis, it was suggested
that dCK, dGK and TK2 belong to one group, which
will be referred to as dNKs, whereas TK1-like enzymes
form a group of their own. This was confirmed by sub-
sequent structure determinations of a multisubstrate
Drosophila melanogaster dNK together with human
dGK [6], followed by human dCK [7], and later on
hTK1 [8,9] and TK from Ureaplasma urealyticum
(Uu-TK) [9]. However, Herpes simplex virus type 1
thymidine kinase shares structural and sequential simi-
larities with dNKs and does not belong to the TK1-
like group of enzymes. dNKs are biological dimers
with overlapping substrate specificity, which can be
attributed to differences of a few residues in the active
site [7,10]. TK1-like enzymes only accept thymidine
and deoxyuridine as substrates, and all interactions
between the substrate and the enzyme are by main-
chain hydrogen bonds to polar groups of the base.
The active site of TK1-like enzymes is smaller than
that found in dNKs and lined with hydrophobic resi-
dues. Whereas TK1-like enzymes have a lasso-domain
which covers the active site when the substrate is
bound, the active site of dNKs is covered by a helical
domain containing an arginine-rich lid. In both
enzyme families, the active site is situated at the C-ter-
minus of the central parallel b-sheet in the a⁄b-
domain, which contains a conserved P-loop
(GXXXGKS ⁄T). Yet another difference between
dNKs and TK1-like enzymes is the presence of a struc-
tural Zn
2+
ion in the lasso-domain of TKs. There are
no structural metals in members of the dNK family.
Furthermore, all TKs that have been structurally
determined form tetramers in the crystals. Enzymes
from the dNK and TK1 family can use different
NTPs, usually prefer ATP as phosphate donor, and
are feedback inhibited by the respective dNTP, such
that dTTP is a feedback inhibitor of TK1-like enzymes
[11]. It can be concluded that, despite structural differ-
ences, dNKs and TKs catalyze the phosphorylation of
deoxyribonucleosides in similar ways [9,12].
In this study, we describe the 3D structure of Ba-TK
in complex with thymidine and a phosphate ion, as well
as Bc-TK with an occupied phosphate donor site. As
these enzymes are essentially identical, these structures
represent the enzyme trapped in different conforma-
tional stages, which reflect structural conformations
that TK adopts along its reaction pathway.
Results
Overall structure
TKs from B. anthracis and B. cereus share 96%
amino-acid sequence identity. Bc-TK consists of 195
amino acids, and Ba-TK is one amino acid shorter.
The last five and the last four amino acids in Bc-TK
and Ba-TK, respectively, are different. Besides the dif-
ferences in the C-termini, there are only three addi-
tional amino acids that are not conserved. The lysine
at position 76 in Ba-TK is a glutamic acid in Bc-TK,
the methionine at position 82 in Ba-TK is a leucine in
Bc-TK, and the alanine at position 147 in Ba-TK is a
valine in Bc-TK. These minor differences do not affect
the overall structures of Ba-TK and Bc-TK, thus these
proteins may be considered structurally identical.
The overall structures of Ba-TK and Bc-TK closely
resemble previously described TK structures [8,9,
13,14]. The enzymes are tetramers with 222-fold sym-
metry and two types of subunit–subunit interaction.
One is formed between helices a1 of two neighboring
subunits, and the other is between the edges of the
b-sheets. The subunit comprises two domains, the
N-terminal a⁄b-domain and the C-terminal lasso-
domain (Fig. 1). The a⁄b-domain is formed from a
central, six-stranded, parallel b-sheet situated between
a long a-helix, a1, and a flexible loop on one side and
three shorter helices, a2–a4, on the other side. We
have chosen to name the flexible loop, which is about
20–25 residues in length (amino acids 46–68 for Ba-TK
and Bc-TK), the phosphate-binding b-hairpin
(P-b-hairpin). As previously described [14], the P-b-
hairpin is a flexible part of the TK, which has been
reported as missing or having a variety of different
conformations. There is also a phosphate-binding
motif, the P-loop (GXXXXGKS ⁄T), in the junction
between b1 and a1 of the a⁄b-domain. The lasso-
domain, so called because of its ability to capture and
position the substrate [9], comprises two perpendicular
b-hairpins, where the longer hairpin opens up to form
a lasso-shaped loop. A Zn
2+
ion ligated by four cys-
teine residues (Cys145, 148, 183 and 186) stabilizes the
lasso-domain. The active site is situated between the
a⁄b-domain and the lasso-domain.
Bacillus thymidine kinase structures U. Kosinska et al.
728 FEBS Journal 274 (2007) 727–737 ª2006 The Authors Journal compilation ª2006 FEBS
Ba-TK
The structure of the Ba-TK–dT complex was refined at
2.7 A
˚resolution to a final R-factor of 20.3% and R
free
of 24.4% (Table 1). There is one subunit in the asym-
metric unit of the space group I4
1
22. Application of
crystallographic symmetries generates the tetramer.
The crystal packing creates a mixed, four-stranded
b-sheet between the tetramers. The N-terminus and
C-terminus from two neighboring subunits of one tetra-
mer form a parallel b-sheet, which is connected in an
antiparallel manner with the N-terminus and C-termi-
nus of a neighboring tetramer. Because of the crystal
contacts, it was possible to trace the entire N-terminus
as well as two residues from the His-tag. At the C-ter-
minus, only the last residue, Arg194, is missing, and
Lys192 and Gln193 have flexible side chains which
lack electron density. Residues 46–62, which are situ-
ated on the P-b-hairpin, also lack electron density and
could not be traced. In previously described TK struc-
tures, this region was reported to have a variety of
conformations or to be missing because of flexibility
[14]. As will be described below, this part of the
enzyme becomes ordered when the phosphate donor
site is occupied.
The Ba-TK–dT complex is very similar to the
Uu-TK–dT complex [14]. The substrate is bound in a
hydrophobic pocket between the a⁄b-domain and the
lasso-domain, surrounded by Phe92, Leu116, Phe120,
Phe125 and Ile170 (Fig. 2A). All hydrogen bonds
between the thymine and the enzyme are to main-chain
atoms such that O2 and N3 form hydrogen bonds to
main-chain atoms of residues in the lasso-domain and
O4 to main-chain atoms in the a⁄b-domain. The
methyl group of thymine points towards Thr155. O3¢
of the deoxyribose makes a hydrogen bond with main-
chain nitrogen of Gly174 in the lasso-domain, and O5¢
is hydrogen-bonded to Glu89, which has been sugges-
ted to be the catalytic base (Fig. 2B) [9].
Besides strong electron density for dT, there is addi-
tional density close to the P-loop which has been inter-
preted as a phosphate ion originating from the
crystallization buffer. The position of the phosphate
corresponds to the c-phosphate of the dTTP molecule
bound as feedback inhibitor to hTK1 and Uu-TK
[8,9]. The phosphate ion is coordinated by the residues
in the P-loop: the side chain of Lys21 and main-chain
atoms of residues 18–20.
Bc-TK
Bc-TK crystallized in the same space group, I4
1
22, as
Ba-TK but with different crystal packing and unit cell
parameters (Table 1). As in Ba-TK crystals, there is
Table 1. Data reduction and refinement statistics. Values in par-
entheses refer to outer resolution shell.
Ba-TK Bc-TK
Space group I4
1
22 I4
1
22
Unit cell parameters (A
˚)a¼b¼73.2
c¼223.7
a¼b¼95.4
c¼204.9
Resolution (A
˚) 2.7 2.8
No. of unique reflections 8799 12035
Multiplicity 14.1 13.6
Completeness (%) 99.9 (99.9) 99.8 (99.8)
R
meas
10.6 (49.3) 10.0 (53.4)
<I⁄rI > 22.6 (5.9) 26.8 (4.2)
Refinement
R(%) 20.3 19.6
R
free
(%) 24.4 23.9
R.m.s.d. bond length (A
˚) 0.011 0.011
R.m.s.d. bond angle () 1.34 1.48
Average Bfactors (A
˚
2
)
a
42.8 57.8
a
Average B factor is calculated for residual B factors.
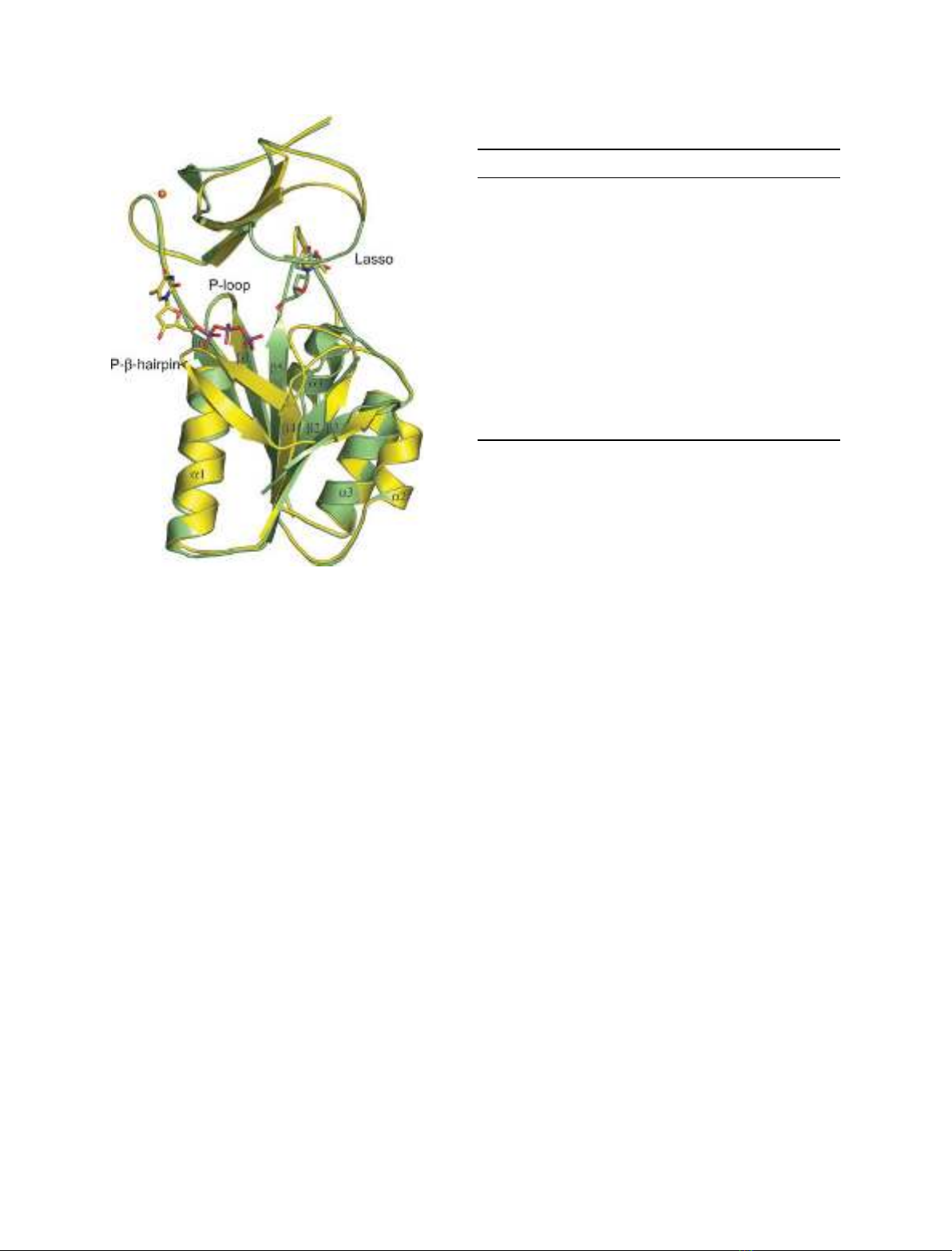
Fig. 1. Superposition of subunits of Ba-TK with dT (in green) and
Bc-TK with phosphate donor-mimicking dTTP and MPD bound in
the thymine-binding pocket (in yellow). The lasso-loop is in closed
conformation when dT is present and in open conformation when
the substrate is absent. The phosphate donor stabilizes the
P-b-hairpin. This part of the molecule is flexible and could not be
traced in Ba-TK.
U. Kosinska et al.Bacillus thymidine kinase structures
FEBS Journal 274 (2007) 727–737 ª2006 The Authors Journal compilation ª2006 FEBS 729

one subunit in the asymmetric unit, thus the tetramer
is formed after application of symmetry operators. The
N-termini of two subunits within the same tetramer
form an antiparallel b-sheet. The crystallographic
interactions between the tetramers involve only the
lasso-domains, which are packed such that the lasso of
one tetramer partly covers the lasso of a crystallo-
graphically related molecule in another tetramer (sup-
plementary Fig. S1). The electron density is continuous
from residue 1 through 191. In contrast with Ba-TK,
the entire region between residue 46 and 62 is fully
traceable, forming a b-hairpin.
The formation of the hairpin is mediated by a nucleo-
tide binding in the phosphate-binding site. Bc-TK was
cocrystallized with the feedback inhibitor dTTP, hence
we expected it to bind as thymine in the substrate-bind-
ing site between the lasso-domain and a⁄b-domain, and
the c-phosphate bound to the P-loop as described previ-
ously [8,9]. Interestingly, there is no electron density for
the inhibitor in the substrate-binding site. Instead, there
is strong positive electron density in the phosphate
donor site, which is situated opposite the substrate-
binding site. It was not possible to conclude from the
initial map whether the electron density represented an
ATP molecule originating from buffers used during
protein purification or a dTTP molecule mimicking a
phosphate donor. Consequently, during the early steps
of ligand fitting, refinement was carried out with both
ATP and dTTP. The electron density corresponding to
the ribose moiety was negative at the 2¢-OH position
when ATP was used in the refinement, and the size of
the electron density for the base was more compatible
with a pyrimidine. From this, we concluded that there
was a deoxyribonucleoside triphosphate, i.e. a dTTP
molecule, occupying the phosphate donor site (Fig. 2B).
dTTP can act as a phosphate donor for Ba-TK, but it
does so poorly compared with ATP: dTTP is only 3%
as efficient as ATP as phosphate donor when dT is used
as substrate [15].
An occupied phosphate donor site gives rise to a
3-A
˚dissociation of subunits interacting by a1. The
base of dTTP is inserted between the a1-helix of two
subunits and is stacked between the rings of Phe18
and Phe34 from the adjacent subunit (Fig. 2B). These
two residues are conserved as hydrophobic residues in
all organisms but Gram-negative bacteria where Phe18
is replaced by asparagine and Phe34 is replaced by
glutamic acid (Fig. 3). The exchange of hydrophobic
residues for polar ones abolishes the hydrophobic
stacking interactions between the base and the enzyme.
The pattern of interaction of ATP with TKs from
Gram-negative bacteria remains to be evaluated. O4 of
the thymine makes a hydrogen bond with the main-
chain nitrogen of Val144. O3¢of deoxyribose is hydro-
gen-bonded to Glu23, and O4¢is hydrogen-bonded to
His58. The phosphates are stabilized by main-chain
nitrogens of P-loop residues as well as by side-chain
interactions with Lys21 and Ser22. In addition to the-
ses interactions, Ser57 and the main-chain nitrogen
from His58, both situated on the P-b-hairpin, also
make hydrogen bonds with the phosphates (Fig. 2B).
The b-phosphate of dTTP as phosphate donor is very
well aligned with the c-phosphate of dTTP bound as a
feedback inhibitor, as observed in Uu-TK and hTK1
[8,9]. dTTP not only provides binding partners for resi-
dues of the P-b-hairpin, but also affects the interac-
tions between subunits of the tetramer.
During the refinement and rebuilding process, posit-
ive density started to appear in the substrate-binding
pocket and was interpreted as a 2-methyl-2,4-pentadiol
(MPD) molecule originating from the crystallization
solution. The position of the MPD molecule
Fig. 2. (A) The active site of Ba-TK is occupied by dT and a phosphate ion. The active site is lined by hydrophobic residues. The map is a
Fo-Fc map contoured at 3r(0.1 e ⁄A
˚
3
). (B) The phosphate donor site of Bc-TK with dTTP mimicking the phosphate donor. The base of the
phosphate donor is stacked between Phe18 and Phe34 each from adjacent subunits shown in yellow and orange, respectively. The phos-
phates are ligated by side-chain and main-chain atoms from the P-loop and P-b-hairpin. The map is a Fo-Fc map contoured at 3r(0.1 e ⁄A
˚
3
).
Bacillus thymidine kinase structures U. Kosinska et al.
730 FEBS Journal 274 (2007) 727–737 ª2006 The Authors Journal compilation ª2006 FEBS

corresponds to the location of thymine of dT or dTTP
as observed in Ba-TK with dT, hTK1 with dTTP, and
Uu-TK with dT or dTTP (Fig. 1). The oxygens of the
MPD molecule form hydrogen bonds to main-chain
and side-chain atoms of the residues in the lasso-loop
(supplementary Fig. S2).
A dNTP molecule can generally bind as a phosphate
donor or a bisubstrate inhibitor. Whether it binds in
one or the other direction is primarily determined by
the affinity of the base of the dNTP for the substrate
site. Normally, the preferred bisubstrate inhibitor is
the dNTP where the base represents the best substrate.
Otherwise, it binds as a phosphate donor. A switch
from the bisubstrate situation to the phosphate donor
situation can be achieved by competing binding in the
substrate site. This was recently shown in a study of
deoxyadenosine kinase, where dCTP could be bound
as a bisubstrate inhibitor in the absence of substrate
but acted as a phosphate donor in the presence of sub-
strate [16]. The high concentration of MPD as a pre-
cipitant in the crystallization, 2.5 m, had some
unexpected consequences. Most surprisingly, it preven-
ted the dTTP molecule from binding in its natural site
as a bisubstrate inhibitor and instead promoted bind-
ing to the phosphate donor site. Although MPD binds
with much lower affinity than dTTP, at this high con-
centration it is able to compete with dTTP, which is
present at about 1000 times lower concentration.
The lasso-loop in Bc-TK has a different conforma-
tion from that observed in Ba-TK (Fig. 1). In Ba-TK,
where dT is occupying the active site, the lasso is
closed down over the active site and stabilized by
hydrogen bonds to thymidine. The absence of a nat-
ural substrate with hydrogen interaction partners, as is
the case in the Bc-TK structure, makes the lasso-loop
flexible. Because of crystallographic interactions, we
were able to trace the entire lasso-loop (supplementary
Fig. S1). An open conformation of the lasso-loop is
also present in the structure of TK from Clostrid-
ium acetobutylicum (Ca-TK) in complex with ADP
(PDB code 1XX6) [17]. In the Ca-TK structure, there
are neither substrates nor crystal contacts that can pro-
vide stabilizing partners. Therefore, parts of the lasso-
loop are missing. The presence of an MPD molecule in
the substrate site of Bc-TK may also add stabilizing
interaction partners for the lasso-loop, but crystal con-
tacts are probably more important for this stabiliza-
tion. Without such, the lasso-loop might have been as
flexible as in Ca-TK.
Subunit–subunit interactions
There are two types of subunit–subunit interaction in
the tetramer. One is between the long a-helices, a1,
between adjacent subunits (Fig. 4A). The helices make
an antiparallel helix pair with hydrophilic or basic
Fig. 3. Amino-acid sequence alignment of the TK1-like enzymes from B. anthracis (AAT57468), B. cereus (DQ384595), C. acetobutylicum
(NP_349490.1), U. urealyticum (NP_078433), human (P04183), mouse (NP_033413), Arabidosis thaliana (AAM63086.1), Escherichia coli
(NP_415754.1) and Yersinia pestis (NP_405720.1). The secondary-structure elements for Ba-TK and Bc-TK are shown above the alignment.
The P-loop and the zinc coordinating motifs are boxed. The catalytic Glu89 is marked in red, and the Phe18 and Phe34, which stack the base
of the phosphate donor, are marked in green. Whereas the catalytic base is conserved among TKs from different kingdoms, the stacking
phenylalanines are exchanged for hydrophilic residues in Gram-negative bacteria. The residues marked in blue take part in subunit–subunit
interactions.
U. Kosinska et al.Bacillus thymidine kinase structures
FEBS Journal 274 (2007) 727–737 ª2006 The Authors Journal compilation ª2006 FEBS 731









![Hình ảnh học bệnh não mạch máu nhỏ: Báo cáo [Năm]](https://cdn.tailieu.vn/images/document/thumbnail/2024/20240705/sanhobien01/135x160/1985290001.jpg)















