
Chapter 7
Dimerization, oligomerization and polymerization of
alkenes and alkynes
The annual production of various polymers can be measured only in
billion tons of which polyolefins alone figure around 100 million tons per
year. In addition to radical and ionic polymerization, a large part of this
huge amount is manufactured by coordination polymerization technology.
The most important Ziegler-Natta, chromium- and metallocene-based
catalysts, however, contain early transition metals which are too oxophilic to
be used in aqueous media. Nevertheless, with the late transition metals there
is some room for coordination polymerization in aqueous systems [1,2] and
the number of studies published on this topic is steadily growing.
7.1 Dimerization and polymerization of ethylene
Coordination polymerization of ethylene by late transition metals is a
rather slow process especially when the catalyst is dissolved in water. In a
study of the interaction of and (tos = tosylate),
both and were
isolated by evaporation of the aqueous phase which had been previously
pressurized with 60 bar ethylene at room temperature for 6 and 18 hours,
respectively. Longer reaction times (72 h) led to the formation of butenes
with no further oligomerization. This aqueous catalytic dimerization was not
selective, the product mixture contained Z-2-butene, E-2-butene and 1-
butene in a 1/2.2/2.2 ratio [3].
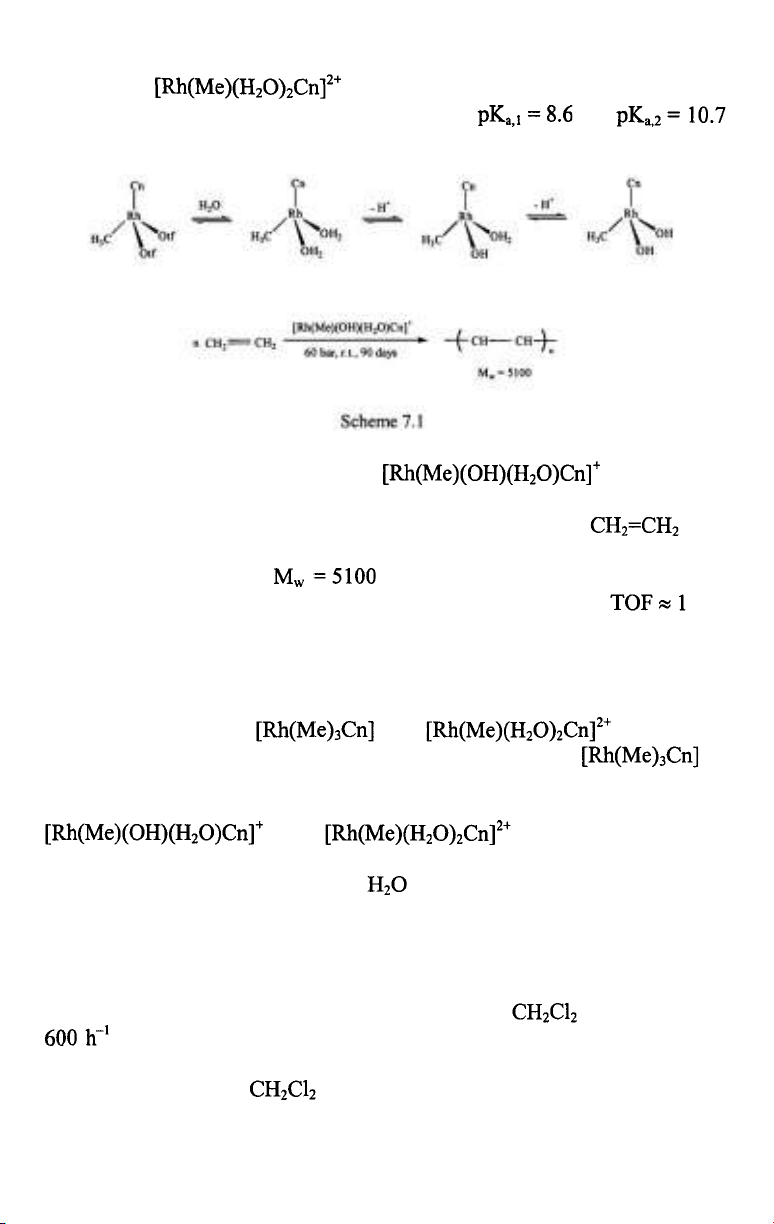
The facially coordinating l,4,7-trimethyl-l,4,7-triazacyclononane (Cn)
ligand forms stable methylrhodium(III) complexes, such as
and (OTf=trifluoromethanesulfonate)
and the latter two have rich aqueous chemistry. When dissolved in water,
readily coordinates two water molecules to form the
237
238
Chapter 7
octahedral in which the aqua ligands undergo
sequential deprotonation in basic solutions with and
(Scheme 7.1) [4].
At 24 °C and 15-60 bar ethylene, catalyzed the
slow polymerization of ethylene [4]. Propylene, methyl acrylate and methyl
methacrylate did not react. After 90 days under 60 bar (the
pressure was held constant throughout) the product was low molecular
weight polyethylene with and a polydispersity index of 1.6. This
is certainly not a practical catalyst for ethylene polymerization ( in a
day), nevertheless the formation and further reactions of the various
intermediates can be followed conveniently which may provide ideas for
further catalyst design. For example, during such investigations it was
established, that only the monohydroxo-monoaqua complex was a catalyst
for this reaction, both and were found
completely ineffective. The lack of catalytic activity of is
understandable since there is no free coordination site for ethylene. Such a
coordination site can be provided by water dissociation from
and and the rate of this
exchange is probably the lowest step of the overall reaction.The hydroxy
ligand facilitates the dissociation of
and this leads to a slow catalysis of
ethene polymerization.
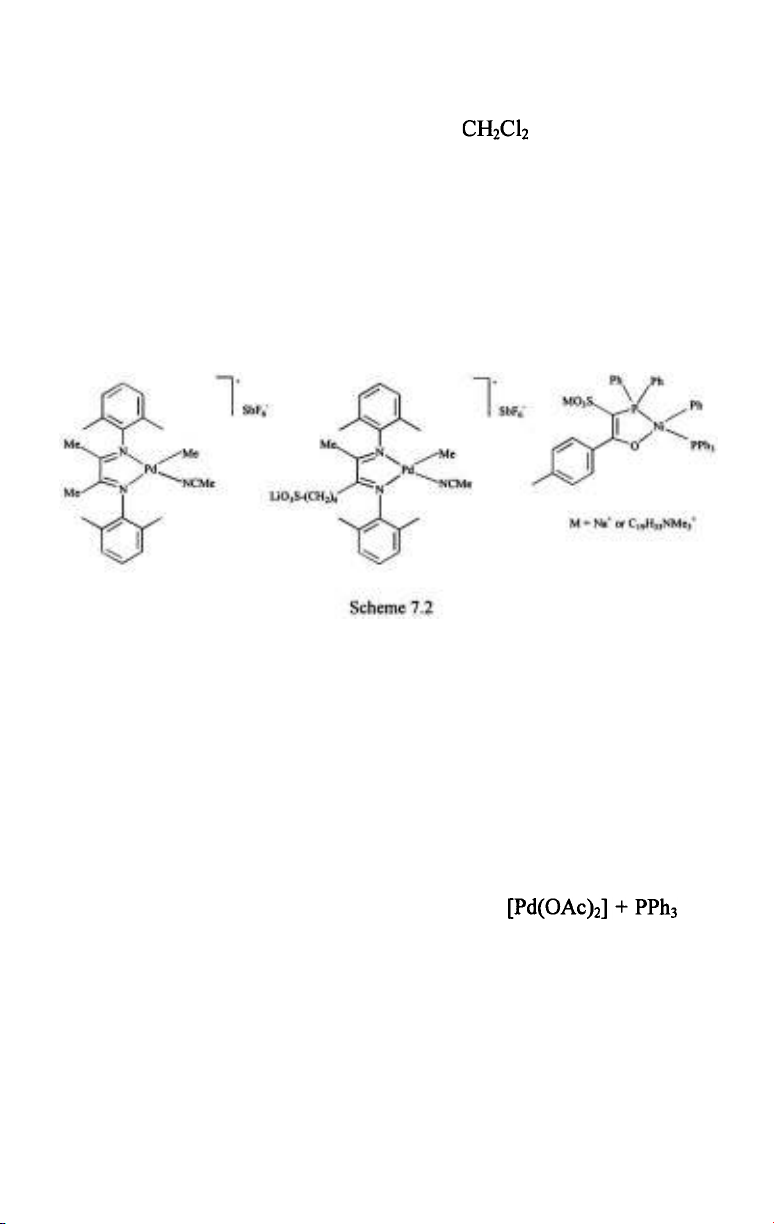
Cationic Pd- and neutral Ni-complexes of chelating N-N or P-O ligands
catalyze the polymerization of ethylene in aqueous media with reasonably
high acitivity (Scheme 7.2) [5,6,61,62]. In fact, the turnover frequencies are
close to those obtained with the same catalysts in (TOF-s 450 vs.
at room temperature). On the other hand, aqueous polymerizations
provided polymers with much higher molecular mass (e.g. 77700 compared
to 14500, obtained in ). The same kind of branching was found in
these polymers, nevertheless the higher molecular mass was manifested in
the physical apperance - the polymers obtained in the aqueous reactions
Dimerization, oligomerization and polymerization of alkenes and
alkynes
239
were rubbery solids while polymerizations in afforded viscous oils.
Very importantly, the active Pd- and Ni-catalysts are water-insoluble,
consequently these aqueous polymerizations were catalyzed by solid
particles of the catalysts suspended in the aqueous phase rather than by
homogeneously dissolved metal complexes. When a palladium catalyst was
made water-soluble by using a sulfoalkyl-modified diimine ligand no
activity whatsoever was observed. The catalytic activity was similarly lost
upon dissolution of the catalysts in the aqueous phase by co-solvents, such
as acetone.
7.2 Telomerization of dienes
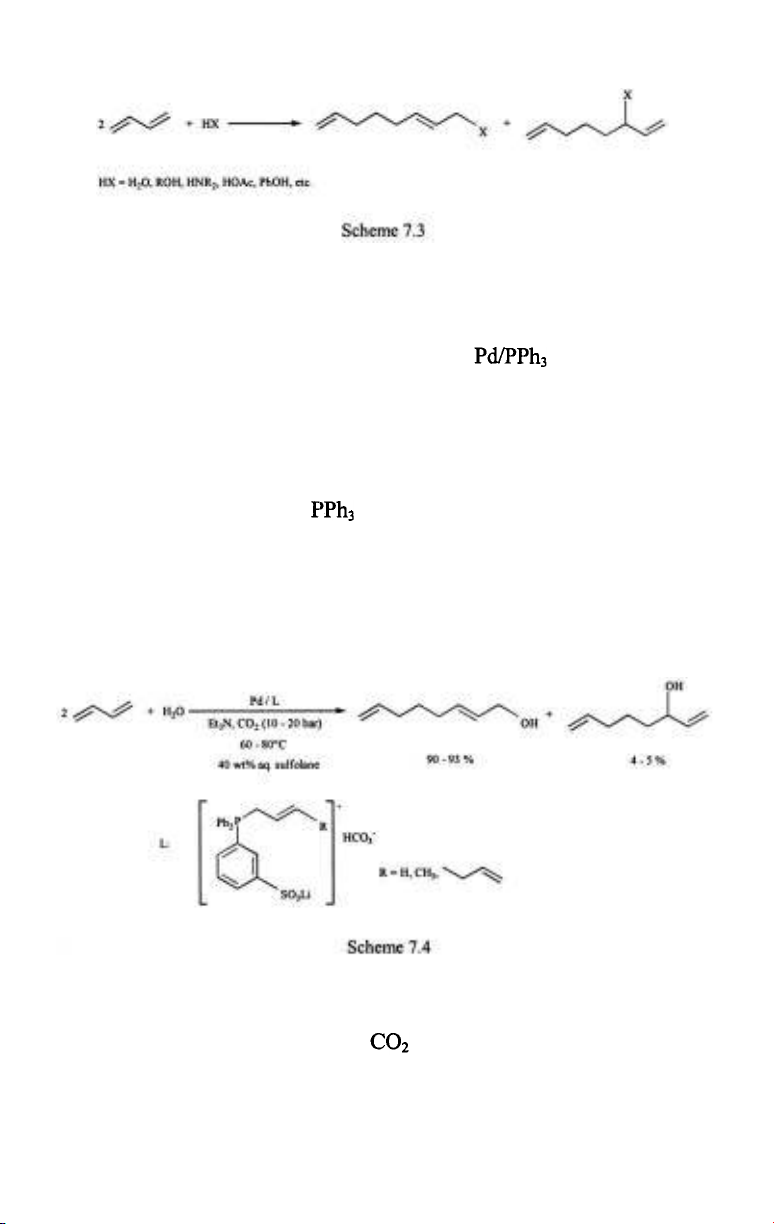
The linear telomerization reaction of dienes was one of the very first
processes catalyzed by water soluble phosphine complexes in aqueous
media [7,8]. The reaction itself is the dimerization of a diene coupled with a
simultaneous nucleophilic addition of HX (water, alcohols, amines,
carboxylic acids, active methylene compounds, etc.) (Scheme 7.3). It is
catalyzed by nickel- and palladium complexes of which palladium catalysts
are substantially more active. In organic solutions gives
the simplest catalyst combination and Ni/TPPTS and Pd/TPPTS were
suggested for running the telomerizations in aqueous/organic biphasic
systems [7]. An aqueous solvent would seem a straightforward choice for
telomerization of dienes with water (the so-called hydrodimerization). In
fact, the possibility of separation of the products and the catalyst without a
need for distillation is a more important reason in this case, too.
240
Chapter 7
The most important aqueous catalytic telomerization reaction is that of
butadiene with water affording octadienols. 2,7-Octadien-1-ol can be easily
hydrogenated to yield 1-octanol, which is used as a raw material for
obtaining phtalate plasticizers for PVC. With or with Pd/TPPTS
this reaction could not be developed into a commercial process due to the
rapid degradation of the catalyst. Such a degradation can be retarded with a
large excess of the respective triarylphosphine, unfortunately this leads to an
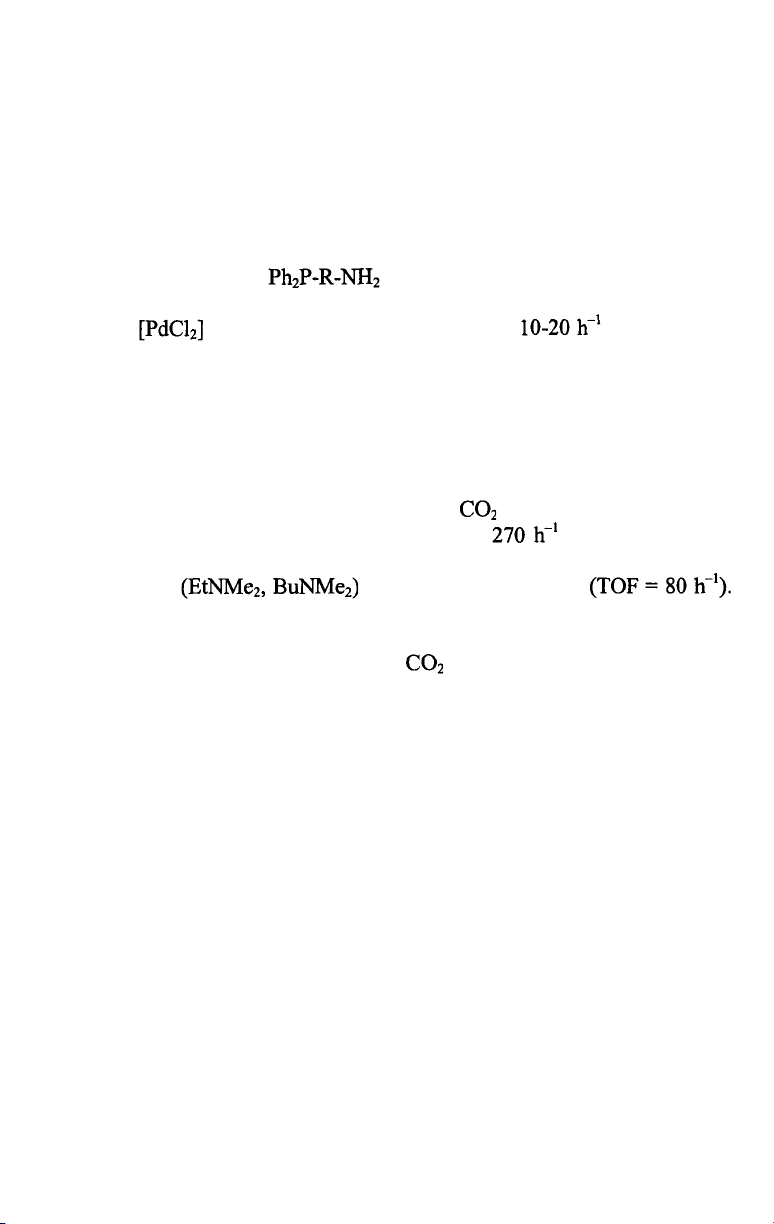
almost complete loss of catalytic activity [9]. This problem was solved by
researchers of Kuraray who introduced the phosphonium salt depicted on
Scheme 7.4 in place of [9-11]. The water-solubility of this
Pd/phosphonium salt catalyst allows to run the hydrodimerization of
butadiene in aqueous/organic two-phase systems. For industrial applications
an aqueous phase containing 40 wt% sulfolane was found the most
advantageous for good reaction rates, easy phase separation during workup
and excellent retainment of the Pd-catalyst.
In the industrial process [12] 1,3-butadiene and water are reacted at 60-
80 °C in an aqueous sulfolane solvent in the presence of triethylamine
hydrogencarbonate under 10-20 bar pressure. The reaction yields linear
telomers mainly, with a 90-93 % selectivity to 2,7-octadien-1-ol together
with 4-5 % l,7-octadien-3-ol. Most of the products are removed from the
reaction mixture by extraction with hexane, and the aqueous sulfolane phase
with the rest of the products, the catalyst and the ammonium bicarbonate is
Dimerization, oligomerization and polymerization of alkenes and
alkynes
241
recycled. The loss of the catalyst is in the range of a few ppm. Based on this
process, Kuraray operates a plant with a capacity of approximately 5000 t/y.
Interestingly, various phosphonium salts have been applied [13] as
constituents of palladium catalysts for hydrodimerization of butadiene and
isoprene about the same time when the results of Kuraray were disclosed.
These were obtained by quaternization of aminoalkylphosphines with
methyl iodide or HCl (
type compounds are known to yield
phosphonium salts with these reagents). Although the catalysts prepared in
situ from were reasonably active (TOF-s of ) the reactions
always yielded complex product mixtures with insufficient selectivity
towards the desired 1,7-octadienyl derivatives.
Aqueous/organic biphasic reaction systems with no co-solvents (such as
the sulfolane above) would be desirable for simplified technologies of diene
telomerization. It was found that with the use of amines which possess one
long alkyl chain, such as dodecyldimethylamine good yields of 2,7-octadien-
catalyst showed high activity with TOF-s up to [14,15]. The main
byproducts were octatrienes and 4-vinylcyclohexene. Amines, which do not
form micelles proved much less useful
The beneficial role of the micelle-forming amines may be in the
solubilization of butadiene in the aqueous phase, furthermore, the
hydrogencarbonate salts formed under pressure may also act as phase
transfer catalysts. This reaction also shows the kinetic complexities of the
telomerization of butadiene with water, the outcome of which greatly
depends on the reaction variables [20].
An interesting application of the palladium-catalyzed telomerizations
is the reaction of butadiene with sucrose (Scheme 7.5) and other
carbohydrates. These substrates are water-soluble therefore it is
straightforward to use an aqueous solvent. The products of this reaction
(mono- and dioctadienylethers) are hydrophobic alkyl glucosides which are
biodegradable, have good surfactant properties and can be used as
emulsifiers in various products. From this respect monoalkylated
carbohydrates are more valuable. The reactions were run in water/organic
solvent (methylisobutylketone, methylethylketone, isopropanol) with a
Pd/TPPTS catalyst in the presence of NaOH. Although selective
monoalkylation could not be achieved, the average number of alkadienyl
chains per carbohydrate unit could be made as low as 1.3 [16]. The products
with an average degree of substitution of 4.7-5.3 are clear, almost clourless
viscous liquids, practically insoluble in water [60]. It is worth mentioning,
that this reaction employs (in part) a renewable raw material and provides a
1-ol could be obtained in water alone, under
pressure. The Pd/TPPTS
![Đề thi kết thúc học phần Nguyên lí Hóa học 2 [mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2025/20251014/anhinhduyet000/135x160/69761760428591.jpg)














