
Hóa học lượng tử Tính Toán
Hóa học lượng tử được sinh ra từ sự toán hóa ngành hóa học bằng Cơ học lượng
tử. Việc áp dụng các phương pháp tính toán vào các vấn đề hóa học dựa trên cơ sở
những tiên đề chính của Cơ học lượng tử, mà nội dung chính của chúng có thể
được tóm tắt như sau:
(i) hàm sóng Ψ(x) của một hạt cơ bản (hay một hệ các hạt cơ bản) chứa đựng mọi
thông tin cần biết liên quan đến hệ đó (trong không gian một chiều). Ý nghĩa vật lý
của hàm số sóng được diễn tả thông qua bình phương của hàm số sóng,
|Ψ(x)(x)|2.dx,
đại lượng này cho biết xác xuất tìm thấy hệ lượng tử trong khoảng không gian (x,
x+dx) được xác định bởi hàm sóng đó;
(ii) mọi tính chất quan sát được, hay đại lượng vật lý đo được, của hệ đều có thể
xác định được từ Ψ(x) thông qua một toán tử tương ứng;
(iii) cho một tính chất hay đại lượng g, một toán tử Ĝ tương ứng được định nghĩa;
áp dụng toán tử Ĝ trên Ψ(x) dẫn đến phương trình trị số riêng Ĝ.Ψ(x) = g.Ψ(x), và
khi giải phương trình này, các trị số riêng g được xác định. Sau khi được chuẩn
hóa, ta nhận được giá trị trung bình cho trị số riêng g trên.
Vào năm 1926, Schrödinger triển khai toán tử cho năng lượng E, được gọi là
Hamiltonian Ĥ, và thành lập phương trình riêng mang tên ông:i
Ĥ Ψ(x) = E Ψ(x) (1)
Trong hóa học, đại lượng quan trọng nhất là năng lượng E của một nguyên tử,
phân tử hay siêu phân tử (gồm những nhân nguyên tử và electron), và sự thay đổi
năng lượng dọc theo tọa độ của phản ứng hóa học. Người làm hóa học cần có các
thông tin này để hiểu diễn biến và cơ chế của phản ứng hóa học dựa trên những
nguyên lý của nhiệt động lực học và động học, và để có thể kiểm soát hay thay đổi
được chúng. Cung cấp thông tin về năng lượng của một hệ phân tử ở mọi trọng
thái electron hay thể loại là một mục đích chính của việc áp dụng những nguyên lý
Cơ học lượng tử vào hóa học. Từ đó đến nay, khởi đi từ những năm đầu của thập
niên 1930, lịch sử của Cơ học lượng tử là một chuỗi dài và liên tục những cố gắng
lớn của nhiều nhà khoa học trong nhiều ngành khoa học (hóa, toán, vật lý, tin học)
nhằm tìm cách giải phương trình (1) để xây dựng hàm số sóng Ψ(x) cho các hệ
phân tử. Phải nói là những “cố gắng lớn” bởi vì toán tử Ĥ cho một phân tử bao

gồm động năng và thế năng của các nhân và electron, cộng với năng lượng tương
tác giữa chúng dẫn đến những tích phân đa tâm phức tạp. Những tích phân này,
nhất là các tích phân trong thế năng tương tác đẩy giữa các electron, dẫn đến việc
không thể giải được chính xác phương trình (1) bằng các phương pháp toán giải
tích cho những hệ đa điện tử.
Song việc áp dụng phương trình Schrödinger đã không bị dừng lại mà đã từng
bước phát triển, đặt nền móng cho ngành Cơ học lượng tử và góp phần mở rộng
cơ sở lý thuyết cho hóa học hiện đại. Những thành tựu của Hóa học lượng tử trong
60 năm qua luôn dựa vào những bước đi trên hai chân. Ngay từ những ngày đầu
cho đến nay, hướng đi chính là phát triển các phương pháp tính thích hợp để xây
dựng nên những hàm số sóng (mà về tính chất là những hàm số sóng gần đúng), so
sánh kết quả với thực nghiệm và tìm cách phát triển lý thuyết để cải thiện. Một
mặt, với mỗi phương pháp mới được đề nghị, năng lượng và những tính chất hóa
học khác tính được từ hàm sóng đã được áp dụng vào các phân tử cụ thể để hiểu
những thông tin và giải thích các hiện tượng hóa học cơ bản nhận được từ thực
nghiệm. Mục đích cuối cùng là vượt qua kết quả từ các con tính trên các phân tử
riêng lẽ để tìm những mô hình và khái niệm chung. Mặt khác, việc cải thiện chất
lượng của Ψ(x) và E luôn được tiếp tục bằng các phương pháp tính toán hoàn thiện
hơn (thường được gọi là “phương pháp cao hơn”) với những con tính luôn phức
tạp hơn nhiều lần, để đạt được những trị số có độ chính xác cao hơn so với thực
nghiệm.
Trên con đường dài này, ngoài kiến thức về chuyên ngành cũng như về các kỹ
thuật tính toán, những chiếc máy tính điện tử luôn có mặt bên cạnh những người
làm Hóa học lượng tử; Máy tính điện tử vừa là công cụ làm việc thân thuộc hàng
ngày, vừa là những người đồng hành tin cậy … Mỗi bước tiến bộ của Hóa học
lượng tử, về phương pháp cũng như độ chính xác, đều gắn liền với một giai đoạn
phát triển mới của khoa học và công nghệ thông tin, hay cụ thể hơn, với một thế hệ
máy tính điện tử.
Trong khuôn khổ của kỷ yếu này (với nhiều độc giả không ở trong ngành hóa học),
chúng tôi không có ý định đi vào chi tiết về phương pháp tính toán phức tạp của
Hóa học lượng tử, mà muốn ghi lại vài nét chấm phá về lịch sử của nó để nêu ra
những đóng góp của các khái niệm lượng tử trong phát triển của hóa học hiện đại.
Áp dụng đầu tiên của phương trình Schrödinger trên nguyên tử hydrogen (H,
nguyên tử nhỏ nhất) dẫn đến khái niệm “orbital nguyên tử” (atomic orbital, viết tắc
là AO), và tiếp theo, việc giải phương trình này cho H2+ (phân tử nhỏ nhất) đưa
đến khái niệm “orbital phân tử” (molecular orbital; mỗi MO là một tổ hợp tuyến

tính của các AO trong phân tử đó). Nhìn lại lịch sử hóa học, orbital là một khái
niệm tuyệt đẹp!. Thật đơn giản và thật cơ bản, vừa định tính, vừa định lượng.
Định tính, vì người làm hóa học có thể xem hình dạng, vị trí, phép đối xứng của
obital để hiểu tính chất vật lý, hóa học và diễn biến của một phản ứng. Có thể hình
dung orbital là một vùng không gian trên phân tử có chứa một cặp electron (vâng,
electron thường kết cặp với trạng thái spin khác nhau; cũng có những chất có
electron tự do). Năng lượng và hình dạng đặc biệt của obital cho phép tiên đoán
được phản ứng của electron khi bị tấn công. Từ đó thật là đơn giản, người làm tổng
hợp hữu cơ dùng bút vẽ nguệch ngoạc trên giấy những obital của các phân tử ban
đầu mà mình đang sử dụng để hiểu được tại sao sau tổng hợp chỉ nhận được chất
này mà không có chất kia. Cũng cùng động tác ấy, người làm hóa lý có thể hiểu
được kết quả của một phổ cực tím vừa ghi được; và dĩ nhiên là các động tác này
đều phải tuân theo những qui luật riêng của chúng (quá nhiều để trình bày chi tiết ở
đây).
Trong giai đoạn phôi thai (1930’s), Linus Pauling đã dùng khái niệm orbital để giải
thích một cách có hệ thống cấu trúc electron của các loại chất hóa học khác nhau,
và từ đó hiểu bản chất của liên kết hóa học (nature of chemical bonding). Giải
thích của Pauling đã thay đổi bộ mặt của hóa học trong nửa sau thế kỷ 20.ii Charles
Coulson dùng thuyết MO để phát triển thêm khái niệm liên kết hóa học và nhất là
khái niệm hóa trị (valence) trong các hợp chất hữu cơ. Robert Mulliken dùng MO
để khảo sát và đưa ra lý thuyết mới về các hợp chất phức phân tử (molecular
complexes) không bền.iii Khái niệm “1 orbital – 2 electron – 3 nguyên tử” trên
nhóm B-H-B do William Lipscomb đề nghị đã cho phép giải thích cấu trúc hình
học của chất boron và đã tạo cơ sở lý thuyết cho lĩnh vực này.iv
Trong giai đoạn đầu (khi chưa có máy tính), Erich Hückel dựa trên các thông tin từ
thực nghiệm đã đưa ra những phép tính lượng tử đơn giản nhằm thay thế cho
nhưng con tính quá phức tạp (không thể tính bằng tay) để xây dựng orbital của các
chất có nối đôi hay các vòng thơm (vì vậy, phương pháp Hückel hay tương tự
thường được gọi là các phương pháp dựa trên kinh nghiệm, empirical methods).
Thế nhưng những con tính HMO (mà sau này dùng làm bài tập cho sinh viên trong
lớp học) đã cho phép giải thích hàng loạt phản ứng hữu cơ, và các phổ cực tím –
ánh sáng thường. Quan trọng hơn, việc có được những orbital Hückel (HMO) cho
những phân tử hữu cơ tương đối lớn đã góp phần cho phép Kenichi Fukui đề nghị
khái niệm “orbital biên” (frontier orbital) vào đầu thập niên 1950’s để giải thích cơ
chế phản ứng hữu cơ, và sau này Roald Hoffmann phát triển thành phương pháp
“Hückel mở rộng” (EHT) để xác định hình dạng và phép đối xứng của những MO

cho các chất tham gia trong một phản ứng tạo vòng. Các kết quả tính toán này của
Hoffmann đưa đến việc hình thành các “qui luật Woodward-Hoffmann” (WH
rules) được công bố vào những năm 1965-1968, và đã tạo nên một dấu ấn lý thuyết
sâu rộng trong sự phát triển của ngành hóa học hữu cơ. Fukui đề nghị rằng: chỉ cần
nhìn vào hình dạng và năng lượng của các orbital biên (HOMO và LUMO) của
một phân tử thì đã có thể hiểu được tính chất và hướng phản ứng của chất đó.
Trong lĩnh vực phổ, một chất hóa học hấp thụ ánh sáng thường hay cực tím là do
một (hay nhiều) electron di chuyển từ một orbital biên này (HOMO) lên obital
khác có năng lượng cao hơn (LUMO). Qui luật WH cũng dựa trên khái niệm này;
song tiên đoán thêm rằng một phản ứng hóa học sẽ dễ xảy ra hơn so với các phản
ứng hóa học khác (xảy ra đồng thời) khi phép đối xứng của các obital biên của nó
được giữ nguyên khi đi từ chất ban đầu đến chất cuối.v
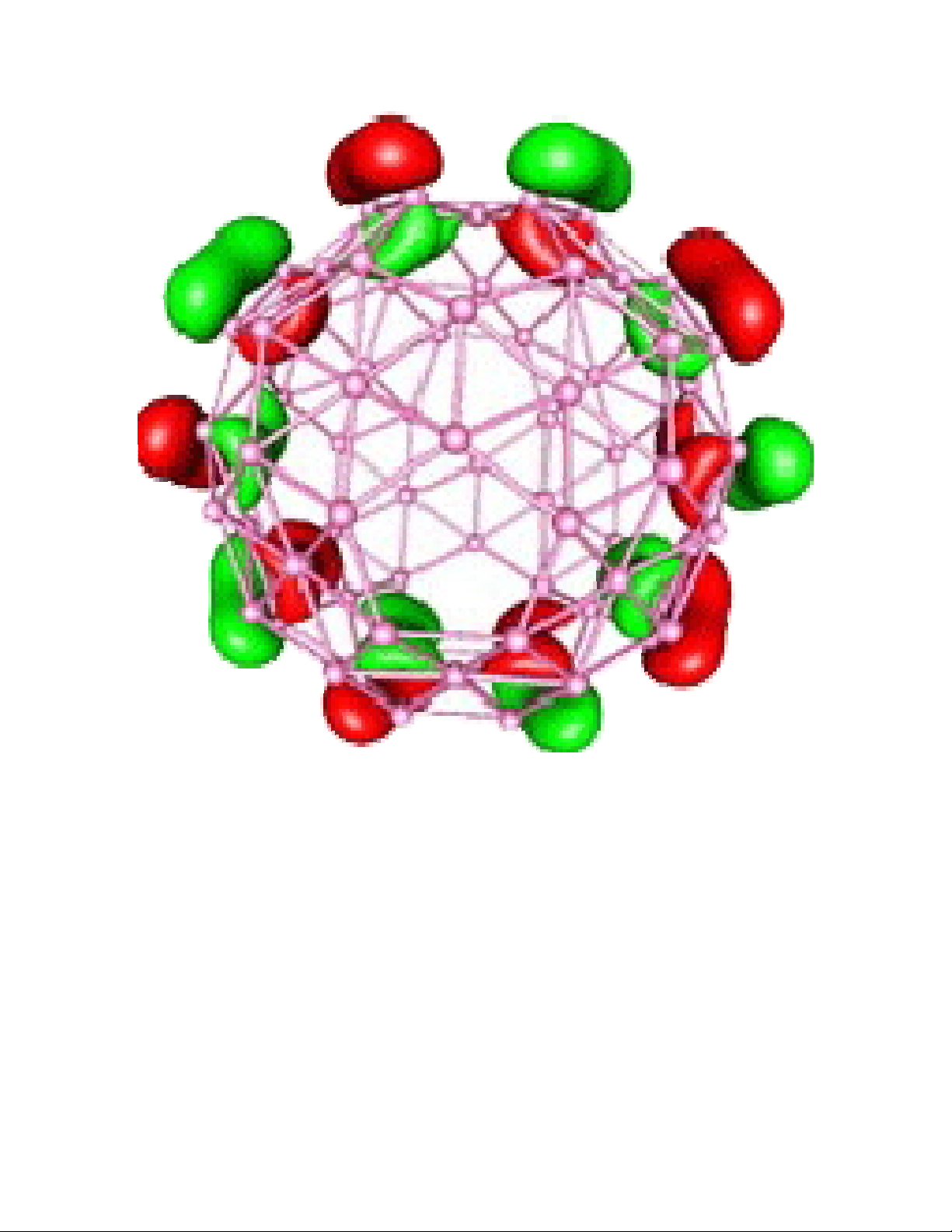
Ví dụ đưa ra ở Hình 1 cho thấy orbital biên HOMO của chất B80. Với 240 electron
hóa trị, chất này được xem là tương đương với hợp chất nổi tiếng C60 (carbon
buckyball). Thật vậy các orbital biên của chất B80 và C60 cũng rất giống nhau
(Hình 1). Hiện nay chất B80 chưa được tổng hợp, song sự tương đương của orbital
biên cho phép tiên đoán chất B80 cũng có những đặc tính của một fullerene (boron
buckyball).vi
Tóm lại, khái niệm MO và quan điểm cho rằng phản ứng hóa học xảy ra chủ yếu là
do mối tương tác giữa orbital của hai chất tham gia phản ứng ban đầu đã thấm vào
tư duy của những người làm hóa học một cách sâu và rộng. Trong nửa sau của thế
kỷ 20, hàng loạt những nguyên lý hóa học mới được ra đời, không những cho các
phân tử hữu cơ, chất khí mà còn cho các phân tử vô cơ, phức kim loại và chất rắn,
và hiện nay là những nanoclusters; và tất cả đều dựa vào thuyết orbital phân tử
(MO). Những khái niệm như các kiểu liên kết hóa học, obital biên, qui luật WH,
siêu liên hợp (hyperconjugation), hay tính thơm (aromaticity)… đã dần dần làm
đầy các chương đầu sách hóa đại cương cho bậc đại học và trung học phổ thông.
B80






















![Đề thi trắc nghiệm giữa kì môn Hóa đại cương 2: [Kèm đáp án/ Tổng hợp đề thi/ Mới nhất]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260514/quangbinhmc2006@gmail.com/135x160/56141778723328.jpg)

![Tài liệu Hóa học đại cương Trường Đại học Công nghệ Giao thông Vận tải [PDF]](https://cdn.tailieu.vn/images/document/thumbnail/2026/20260511/vispacex_27/135x160/7711778501472.jpg)

%20--%3e%3cdefs%3e%3cstyle%3e%20.st0%20{%20fill:%20%23fff;%20}%20.st1%20{%20fill:%20%237800fa;%20}%20%3c/style%3e%3c/defs%3e%3cpath%20class='st1'%20d='M117.78,12.18H43.11c2.9,3.47,4.65,7.94,4.65,12.82,0,5.6-2.3,10.66-6.01,14.29h76.02l7.22-13.56-7.22-13.56Z'/%3e%3cg%3e%3cpath%20class='st0'%20d='M53.58,26.17h-.59v-1.46h.59v-4.96h2.83c1.78,0,2.67.94,2.67,2.82v5.76c0,1.87-.89,2.81-2.67,2.81h-2.83v-4.96ZM55.36,21.37v3.34h1.1v1.46h-1.1v3.34h1.01c.61,0,.91-.37.91-1.1v-5.93c0-.74-.3-1.1-.91-1.1h-1.01Z'/%3e%3cpath%20class='st0'%20d='M65.99,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM65.28,18.04c-.25.46-.51.77-.75.94-.21.15-.47.22-.79.22-.26,0-.57-.07-.92-.22l-.38-.15c-.14-.05-.26-.07-.37-.07-.3,0-.53.18-.71.54l-.91-.68c.25-.46.51-.77.75-.94.21-.14.48-.21.79-.21.26,0,.57.07.92.21l.38.15c.14.05.26.07.37.07.3,0,.53-.18.71-.54l.91.68ZM61.91,27.52h1.73l-.87-5.76-.87,5.76Z'/%3e%3cpath%20class='st0'%20d='M74.53,26.89v1.52c0,1.91-.89,2.86-2.67,2.86s-2.67-.95-2.67-2.86v-5.93c0-1.91.89-2.86,2.67-2.86s2.67.95,2.67,2.86v1.11h-1.69v-1.22c0-.75-.31-1.12-.93-1.12s-.93.37-.93,1.12v6.15c0,.74.31,1.11.93,1.11s.93-.37.93-1.11v-1.63h1.69Z'/%3e%3cpath%20class='st0'%20d='M81.4,31.14h-1.8l-.31-2.07h-2.19l-.31,2.07h-1.64l1.82-11.39h2.62l1.82,11.39ZM75.9,19.2l1.52-1.91h1.71l1.51,1.91h-1.61l-.76-.95-.75.95h-1.61ZM77.32,27.52h1.73l-.87-5.76-.87,5.76ZM83.1,15.99l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M84.86,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM84.01,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M93.51,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM92.66,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3cpath%20class='st0'%20d='M98.8,31.14h-1.79v-11.39h1.79v4.88h2.03v-4.88h1.83v11.39h-1.83v-4.88h-2.03v4.88Z'/%3e%3cpath%20class='st0'%20d='M105.36,24.55h2.46v1.62h-2.46v3.34h3.09v1.63h-4.88v-11.39h4.88v1.63h-3.09v3.18ZM108.17,17.29l-1.76,1.91h-1.26l1.17-1.91h1.86Z'/%3e%3cpath%20class='st0'%20d='M112.2,19.75c1.78,0,2.67.94,2.67,2.82v1.48c0,1.87-.89,2.81-2.67,2.81h-.85v4.28h-1.79v-11.39h2.64ZM111.35,21.37v3.86h.85c.58,0,.87-.36.87-1.08v-1.71c0-.71-.29-1.07-.87-1.07h-.85Z'/%3e%3c/g%3e%3ccircle%20class='st1'%20cx='25'%20cy='25'%20r='20'/%3e%3cpath%20class='st0'%20d='M32.78,19.27c2.92,0,4.43,2.55,5.28,5.33l.71,2.17c.14.38-.33.75-.71.75h-5.61c.19-.33.24-.71.09-1.08l-.75-2.45c-.43-1.32-.99-2.64-1.79-3.77.75-.57,1.65-.94,2.78-.94h0ZM25,18.38c3.25,0,4.9,2.78,5.89,5.89l.76,2.45c.14.42-.33.8-.8.8h-11.69c-.42,0-.94-.38-.8-.8l.75-2.45c.99-3.11,2.64-5.89,5.89-5.89h0ZM25,11.35c1.74,0,3.11,1.37,3.11,3.11s-1.37,3.11-3.11,3.11-3.11-1.41-3.11-3.11,1.41-3.11,3.11-3.11h0ZM17.27,19.27c1.08,0,1.98.38,2.73.94-.8,1.13-1.37,2.45-1.74,3.77l-.8,2.45c-.14.38-.05.75.09,1.08h-5.56c-.42,0-.9-.38-.75-.75l.71-2.17c.9-2.78,2.41-5.33,5.33-5.33h0ZM17.27,12.91c1.51,0,2.78,1.27,2.78,2.83s-1.27,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM32.78,12.91c1.56,0,2.78,1.27,2.78,2.83s-1.23,2.83-2.78,2.83-2.83-1.27-2.83-2.83,1.27-2.83,2.83-2.83h0ZM27.07,28.56v.09c0,.57-.24,1.08-.61,1.46h0v.05c-.38.33-.9.57-1.46.57s-1.08-.24-1.46-.61h0c-.38-.38-.61-.9-.61-1.46v-.09h1.41v.09c0,.19.05.38.19.47v.05c.09.09.28.19.47.19s.38-.09.47-.19v-.05c.14-.09.24-.28.24-.47t-.05-.09h1.41ZM30.99,28.56v.09c0,1.65-.66,3.16-1.74,4.24-1.08,1.08-2.59,1.79-4.24,1.79s-3.16-.71-4.24-1.79l-.05-.05c-1.04-1.08-1.7-2.55-1.7-4.2v-.09h1.41v.09c0,1.27.47,2.4,1.27,3.25h.05c.85.85,1.98,1.37,3.25,1.37s2.4-.52,3.25-1.37c.85-.8,1.37-1.98,1.37-3.25v-.09h1.37ZM34.99,28.56v.09c0,2.78-1.13,5.28-2.92,7.07-1.79,1.79-4.29,2.92-7.07,2.92s-5.23-1.13-7.07-2.92c-1.79-1.79-2.92-4.29-2.92-7.07v-.09h1.41v.09c0,2.4.94,4.53,2.5,6.08,1.56,1.56,3.72,2.5,6.08,2.5s4.52-.94,6.08-2.5c1.56-1.56,2.5-3.68,2.5-6.08v-.09h1.41Z'/%3e%3c/svg%3e)